下面是我曾经接手过的一个项目,内容是个人的经验,解释也不一定完全正确,大家参考了!!!
小改进,大成功--药材TLC鉴别实例
关于××××丸成品鉴别方法的改进
原有方法:
(1)取本品××g,加乙醚20ml,超声处理5分钟,滤过,药渣及滤纸一并置烧瓶中,加7%硫酸乙醇-水(1:3)混合液20ml,加热回流3小时,放冷,用氯仿振摇提取二次,每次20ml,合并氯仿液,加水30ml洗涤,弃去洗液,氯仿液用无水硫酸钠脱水,滤过,滤液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取桔梗对照药材1g,同法制成对照药材溶液。照薄层色谱法(中国药典2005年版一部附录VIB)试验,吸取上述两种溶液各1~2µl,分别点于同一硅胶G薄层板上,以氯仿-乙醚(1:1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色斑点。
(2)取甘草次酸对照品,加甲醇制成每1 ml含1mg的溶液,作为对照品溶液。照薄层色谱法(中国药典2005年版一部附录VIB)试验,吸取[鉴别](1)项下的供试品溶液8µl、对照品溶液5µl,分别点于同一硅胶GF254薄层板上,以石油醚(30~60℃)-氯仿-冰醋酸(10:5:1)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。
改进方法:
(1)取本品××g,加乙醚20ml,超声处理5分钟,滤过,药渣及滤纸再加氯仿30ml,超声10分钟,离心,药渣置烧瓶中,加氯仿30ml和盐酸2ml,加热回流1小时,滤过,滤液蒸干,残渣加无水乙醇1ml使溶解,作为供试品溶液。另取桔梗对照药材0.25g,同法制成对照药材溶液。照薄层色谱法(中国药典2005年版一部附录VIB)试验,吸取上述两种溶液各2~5µl,分别点于同一硅胶G薄层板上,以氯仿-乙醚(1:1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色斑点。
(2)取甘草次酸对照品,加甲醇制成每1 ml含1mg的溶液,作为对照品溶液。照薄层色谱法(中国药典2005年版一部附录VIB)试验,吸取[鉴别](1)项下的供试品溶液和对照品溶液各2~5µl,分别点于同一硅胶GF254薄层板上,以石油醚(30~60℃)-氯仿-冰醋酸(10:5:1)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。
关于××××丸鉴别方法的改进的说明
原有方法对于样品和对照药材的提取存在着一定的问题,具体表现在如下几个方面:
(1)对于甘草次酸的鉴别的机理说明:
A乙醚的超声处理的目的:除去药材或浸膏中的酯溶性成分和制剂中大量存在的水溶性辅料,这些物质的存在直接影响薄层鉴别;
B氯仿的超声处理的目的:除去药材、浸膏和制剂中的甘草次酸成分;
C酸解的目的:通过酸解,可以使药材、浸膏和制剂中的甘草酸水解成为甘草次酸成分,这样可以与对照品进行比较;
原有方法存在的不足:
A加入酸的量不足,使得甘草酸水解不充分,只有少量甘草酸发生水解,致使该方法的重现性很差;
B水解时间长,不适合企业的生产实际需要;
C由于甘草次酸在氯仿和水中都有一定的溶解能力,无水硫酸钠脱水也可能包裹一定量的甘草次酸,所以原方法用氯仿萃取二次、加水30ml洗涤、无水硫酸钠脱水三项操作使得鉴别结果重现性很差,而且这些操作很费时,不同人员的操作的差异比较大。
(2)对于桔梗的鉴别的机理说明:
A乙醚的超声处理的目的:除去药材或浸膏中的酯溶性成分(如脂肪酸、脂肪油等)和制剂中大量存在的水溶性辅料,这些物质的存在直接影响薄层鉴别;
B氯仿的超声处理的目的:除去药材、浸膏和制剂中的桔梗皂苷成分;
C酸解的目的:通过酸解,可以使药材、浸膏和制剂中的桔梗皂苷(包括桔梗皂苷A、B、D2、D3,远志苷D、D2,桔梗皂酸A甲酯等)水解成为苷元(包括桔梗皂苷元、远志酸,桔梗酸等)成分,这样可以与对照药材进行比较;
原有方法存在的不足:
A加入酸的量不足,使得桔梗皂苷水解不充分,只有少量桔梗皂苷发生水解,致使该方法的重现性很差;
B水解时间长,不适合企业的生产实际需要;
C由于甘草次酸在氯仿和水中都有一定的溶解能力,无水硫酸钠脱水也可能包裹一定量的甘草次酸,所以原方法用氯仿萃取二次、加水30ml洗涤、无水硫酸钠脱水三项操作使得鉴别结果重现性很差,而且这些操作很费时,不同人员的操作的差异比较大。
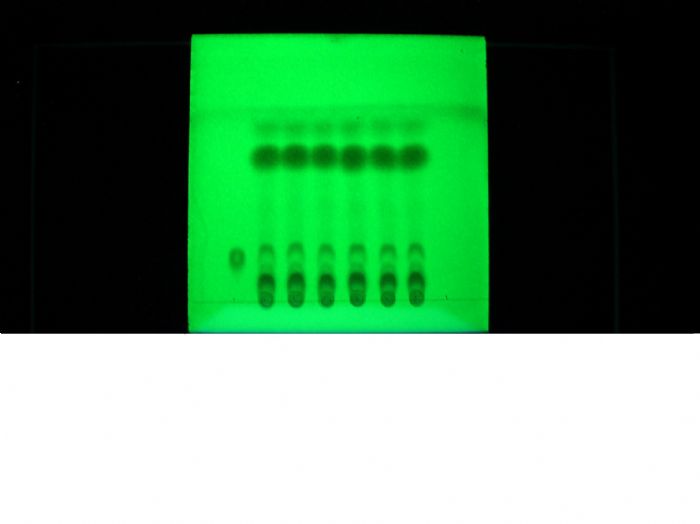
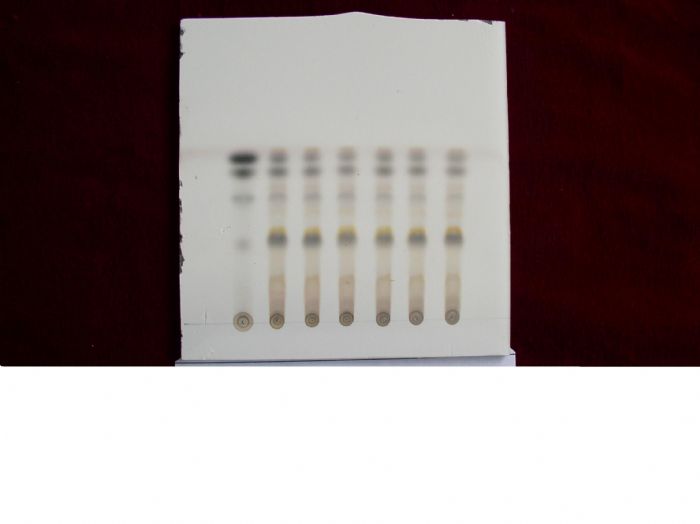
TLC图片!
1、第一张是甘草鉴别,第一个样点为对照品样点,其它为不同批次产品样点。
2、第二张是桔梗鉴别,第一个样点为对照药材样点,其它为不同批次产品样点。
3、第三、四张是以前试验的记录,由于薄层板太差所以电子图片就没有留下,只留下记录~