小中大回复 #6 aa_tang 的帖子
感谢aa_tang,分析得很全面,也很有道理。
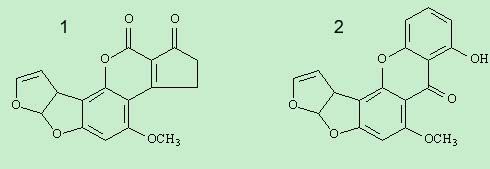
我的目的其实是关注一个代谢途径的所有前体化合物和代谢终产物,它们的结构比较相似,都能溶解于丙酮,甲醇,应该属于中等极性化合物。虽然结构相似,但在上述条件下的保留时间相差比较大。比如化合物1和2(结构式见附件)的Rt分别为18.78min和34.38min(从结构式判断,我认为化合物2的极性应该大一些,但是结果正好相反,难道化合物2属于脂溶性化合物?)。所以其它化合物的极性相对大小也不能贸然判断。这就给样品提取和净化方法带来了难度。进行样品纯化又担心会损失目标化合物,不纯化又出现馒头峰。
关于您的建议,还有几个问题。
1. 我用的是aglient的RRLC系统,色谱柱也是配套的粒径1.8um的,相当于waters的UPLC了,但是结果你也看到了。
2. 您建议流动相用5-100%乙腈,60min内梯度洗脱,那么 在30min-60min内,乙腈的比例从50%-100%线性变化。
按照我采用的条件,在30-50min内也是乙腈含量50-100%。
这两个条件基本上变化不大吧。
感谢aa_tang能耐心看完,还能再给一些建议吗?谢谢!
2008-12-11 14:13
 1.jpg (21.23 KB)
1.jpg (21.23 KB)