 图片附件: 93908-2.0-embed.jpg (2012-9-19 16:54, 109.36 KB) / 该附件被下载次数 6
图片附件: 93908-2.0-embed.jpg (2012-9-19 16:54, 109.36 KB) / 该附件被下载次数 6 图片附件: 127193-cell1200-embed.JPG (2012-9-21 12:26, 17.32 KB) / 该附件被下载次数 29
图片附件: 127193-cell1200-embed.JPG (2012-9-21 12:26, 17.32 KB) / 该附件被下载次数 29 图片附件: 31915911.jpg (2012-9-21 16:26, 37.27 KB) / 该附件被下载次数 7
图片附件: 31915911.jpg (2012-9-21 16:26, 37.27 KB) / 该附件被下载次数 7 图片附件: 79015009.jpg (2012-9-21 16:27, 56.47 KB) / 该附件被下载次数 8
图片附件: 79015009.jpg (2012-9-21 16:27, 56.47 KB) / 该附件被下载次数 8 图片附件: 53903832.snap.jpg (2012-9-21 16:31, 54.15 KB) / 该附件被下载次数 9
图片附件: 53903832.snap.jpg (2012-9-21 16:31, 54.15 KB) / 该附件被下载次数 9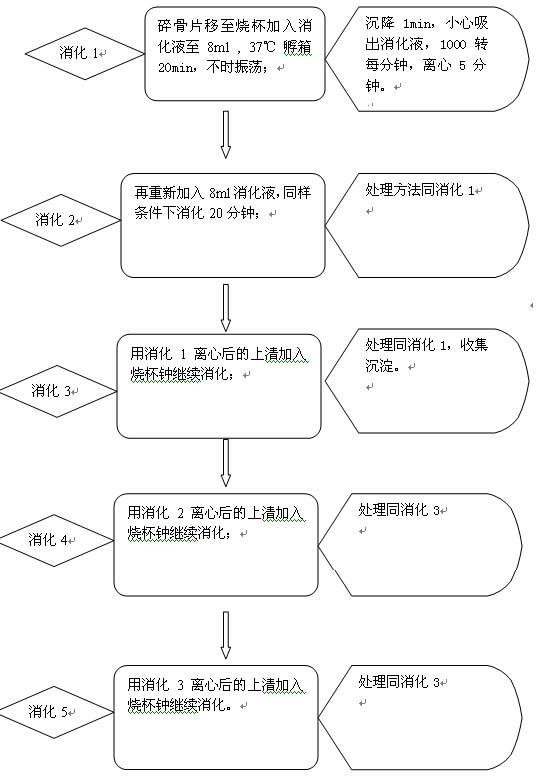 图片附件: 88012291.gif (2012-9-22 15:09, 45.5 KB) / 该附件被下载次数 8
图片附件: 88012291.gif (2012-9-22 15:09, 45.5 KB) / 该附件被下载次数 8 图片附件: 40322391.gif (2012-9-22 15:10, 49.47 KB) / 该附件被下载次数 23
图片附件: 40322391.gif (2012-9-22 15:10, 49.47 KB) / 该附件被下载次数 23 图片附件: 56354277_snap.jpg (2012-9-27 10:08, 56.12 KB) / 该附件被下载次数 34
图片附件: 56354277_snap.jpg (2012-9-27 10:08, 56.12 KB) / 该附件被下载次数 34
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |