Chromatin Immunoprecipitation(ChIP)实验指南及技术总结
 图片附件: 14958764.jpg (2013-4-16 12:35, 3.54 KB) / 该附件被下载次数 46
图片附件: 14958764.jpg (2013-4-16 12:35, 3.54 KB) / 该附件被下载次数 46 图片附件: 99322194.jpg (2013-4-16 12:35, 5.39 KB) / 该附件被下载次数 37
图片附件: 99322194.jpg (2013-4-16 12:35, 5.39 KB) / 该附件被下载次数 37 图片附件: 93505325.snap.jpg (2013-4-16 14:33, 123.87 KB) / 该附件被下载次数 23
图片附件: 93505325.snap.jpg (2013-4-16 14:33, 123.87 KB) / 该附件被下载次数 23
 图片附件: 34731475.png (2013-4-16 15:45, 43.74 KB) / 该附件被下载次数 32
图片附件: 34731475.png (2013-4-16 15:45, 43.74 KB) / 该附件被下载次数 32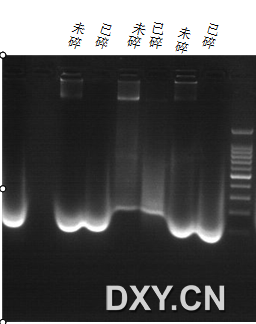 图片附件: 12840390.png (2013-4-16 16:20, 22.82 KB) / 该附件被下载次数 70
图片附件: 12840390.png (2013-4-16 16:20, 22.82 KB) / 该附件被下载次数 70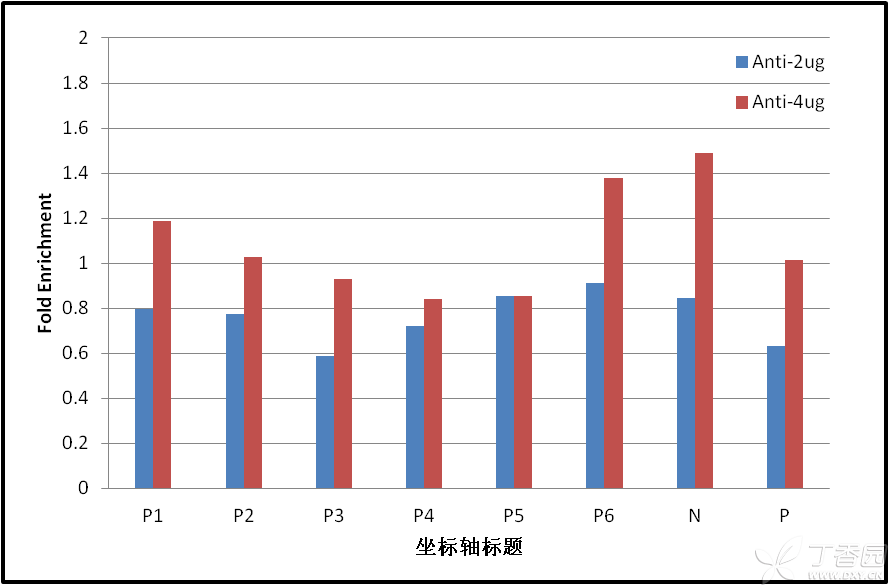
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |