 图片附件: 70838749.jpg (2013-4-19 13:20, 14.1 KB) / 该附件被下载次数 33
图片附件: 70838749.jpg (2013-4-19 13:20, 14.1 KB) / 该附件被下载次数 33 图片附件: 40837764.jpg (2013-4-19 13:20, 51.79 KB) / 该附件被下载次数 33
图片附件: 40837764.jpg (2013-4-19 13:20, 51.79 KB) / 该附件被下载次数 33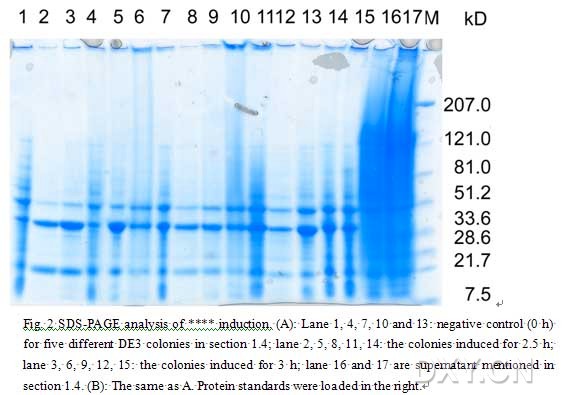 图片附件: 30859696.snap.jpg (2013-4-19 13:27, 50.45 KB) / 该附件被下载次数 31
图片附件: 30859696.snap.jpg (2013-4-19 13:27, 50.45 KB) / 该附件被下载次数 31 图片附件: 92803121.jpg (2013-4-19 13:27, 29.45 KB) / 该附件被下载次数 28
图片附件: 92803121.jpg (2013-4-19 13:27, 29.45 KB) / 该附件被下载次数 28 图片附件: 58751685.jpg (2013-4-20 10:09, 42.72 KB) / 该附件被下载次数 11
图片附件: 58751685.jpg (2013-4-20 10:09, 42.72 KB) / 该附件被下载次数 11 图片附件: 83427493.jpg (2013-4-20 10:18, 32.55 KB) / 该附件被下载次数 4
图片附件: 83427493.jpg (2013-4-20 10:18, 32.55 KB) / 该附件被下载次数 4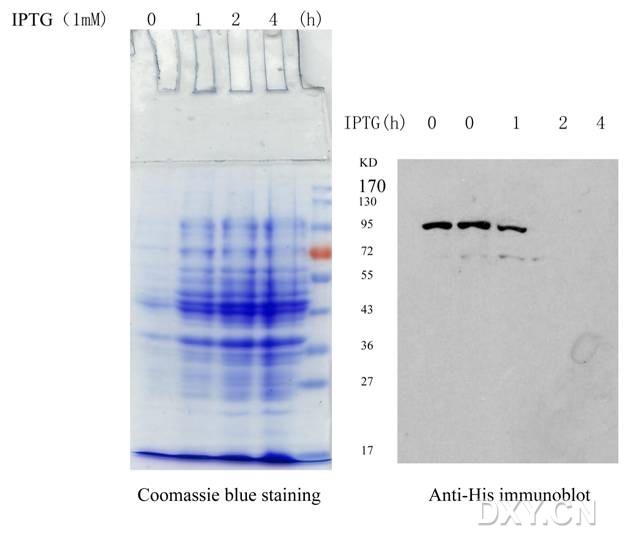 图片附件: 32512283.jpg (2013-4-20 10:35, 14.71 KB) / 该附件被下载次数 7
图片附件: 32512283.jpg (2013-4-20 10:35, 14.71 KB) / 该附件被下载次数 7 图片附件: 32512283.jpg (2013-4-20 10:36, 14.71 KB) / 该附件被下载次数 8
图片附件: 32512283.jpg (2013-4-20 10:36, 14.71 KB) / 该附件被下载次数 8
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |