 图片附件: 58204867.png (2013-5-2 13:35, 12.36 KB) / 该附件被下载次数 20
图片附件: 58204867.png (2013-5-2 13:35, 12.36 KB) / 该附件被下载次数 20 图片附件: 94737148.snap.jpg (2013-5-2 13:38, 23.77 KB) / 该附件被下载次数 19
图片附件: 94737148.snap.jpg (2013-5-2 13:38, 23.77 KB) / 该附件被下载次数 19 图片附件: 56473001.png (2013-5-2 13:38, 59.63 KB) / 该附件被下载次数 29
图片附件: 56473001.png (2013-5-2 13:38, 59.63 KB) / 该附件被下载次数 29 图片附件: 37674285.snap.jpg (2013-5-2 13:38, 26.77 KB) / 该附件被下载次数 2
图片附件: 37674285.snap.jpg (2013-5-2 13:38, 26.77 KB) / 该附件被下载次数 2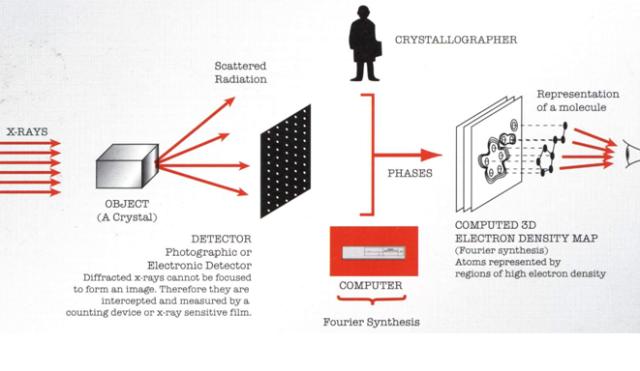 图片附件: 91993372.jpg (2013-5-2 13:39, 73.34 KB) / 该附件被下载次数 10
图片附件: 91993372.jpg (2013-5-2 13:39, 73.34 KB) / 该附件被下载次数 10 图片附件: 58007745.png (2013-5-2 13:40, 54.61 KB) / 该附件被下载次数 12
图片附件: 58007745.png (2013-5-2 13:40, 54.61 KB) / 该附件被下载次数 12 图片附件: 24227511.png (2013-5-2 13:41, 39.98 KB) / 该附件被下载次数 8
图片附件: 24227511.png (2013-5-2 13:41, 39.98 KB) / 该附件被下载次数 8 图片附件: 44722803.png (2013-5-2 13:41, 6.55 KB) / 该附件被下载次数 9
图片附件: 44722803.png (2013-5-2 13:41, 6.55 KB) / 该附件被下载次数 9 图片附件: 82587016.snap.jpg (2013-5-2 13:41, 31.4 KB) / 该附件被下载次数 8
图片附件: 82587016.snap.jpg (2013-5-2 13:41, 31.4 KB) / 该附件被下载次数 8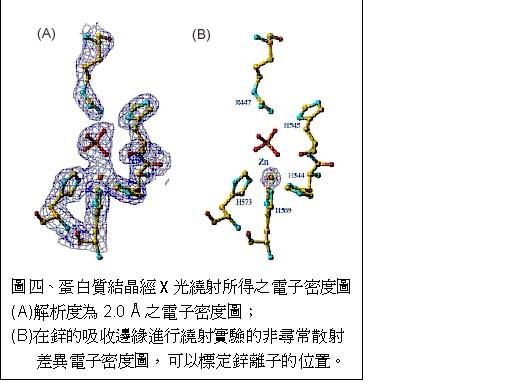 图片附件: 66728937.png (2013-5-2 13:42, 6.91 KB) / 该附件被下载次数 12
图片附件: 66728937.png (2013-5-2 13:42, 6.91 KB) / 该附件被下载次数 12
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |