 图片附件: 71750349.snap.jpg (2013-5-14 12:40, 17.72 KB) / 该附件被下载次数 16
图片附件: 71750349.snap.jpg (2013-5-14 12:40, 17.72 KB) / 该附件被下载次数 16 图片附件: 64481768.snap.jpg (2013-5-14 12:48, 19.84 KB) / 该附件被下载次数 20
图片附件: 64481768.snap.jpg (2013-5-14 12:48, 19.84 KB) / 该附件被下载次数 20 图片附件: 51334328.snap.jpg (2013-5-14 13:25, 57.82 KB) / 该附件被下载次数 26
图片附件: 51334328.snap.jpg (2013-5-14 13:25, 57.82 KB) / 该附件被下载次数 26 图片附件: 46398015.jpg (2013-5-14 13:34, 7.63 KB) / 该附件被下载次数 13
图片附件: 46398015.jpg (2013-5-14 13:34, 7.63 KB) / 该附件被下载次数 13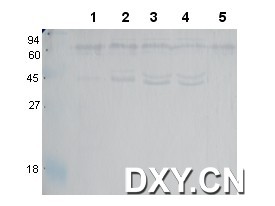 图片附件: 85347431.gif (2013-5-14 13:34, 1.78 KB) / 该附件被下载次数 22
图片附件: 85347431.gif (2013-5-14 13:34, 1.78 KB) / 该附件被下载次数 22
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |