 图片附件: 60252517.snap.jpg (2013-9-5 17:38, 26.72 KB) / 该附件被下载次数 14
图片附件: 60252517.snap.jpg (2013-9-5 17:38, 26.72 KB) / 该附件被下载次数 14 图片附件: 16986602.jpg (2013-9-6 15:59, 29.28 KB) / 该附件被下载次数 22
图片附件: 16986602.jpg (2013-9-6 15:59, 29.28 KB) / 该附件被下载次数 22 图片附件: 91018831.snap.jpg (2013-9-6 16:35, 4.8 KB) / 该附件被下载次数 7
图片附件: 91018831.snap.jpg (2013-9-6 16:35, 4.8 KB) / 该附件被下载次数 7 图片附件: 99592421.jpg (2013-9-6 16:57, 14.56 KB) / 该附件被下载次数 18
图片附件: 99592421.jpg (2013-9-6 16:57, 14.56 KB) / 该附件被下载次数 18 图片附件: 64777716.snap.jpg (2013-9-9 13:55, 97.42 KB) / 该附件被下载次数 16
图片附件: 64777716.snap.jpg (2013-9-9 13:55, 97.42 KB) / 该附件被下载次数 16 图片附件: 51084069.jpg (2013-9-9 16:32, 13.97 KB) / 该附件被下载次数 16
图片附件: 51084069.jpg (2013-9-9 16:32, 13.97 KB) / 该附件被下载次数 16 图片附件: 90394113.snap.jpg (2013-9-10 13:34, 53.67 KB) / 该附件被下载次数 24
图片附件: 90394113.snap.jpg (2013-9-10 13:34, 53.67 KB) / 该附件被下载次数 24 图片附件: 47502086.snap.jpg (2013-9-10 13:38, 12.2 KB) / 该附件被下载次数 25
图片附件: 47502086.snap.jpg (2013-9-10 13:38, 12.2 KB) / 该附件被下载次数 25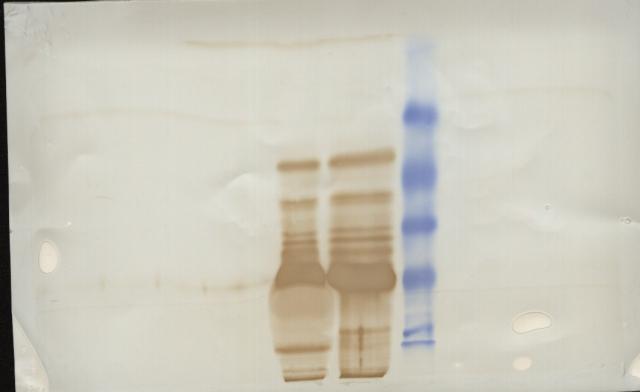 图片附件: 40663180.snap.jpg (2013-9-10 13:43, 28.95 KB) / 该附件被下载次数 23
图片附件: 40663180.snap.jpg (2013-9-10 13:43, 28.95 KB) / 该附件被下载次数 23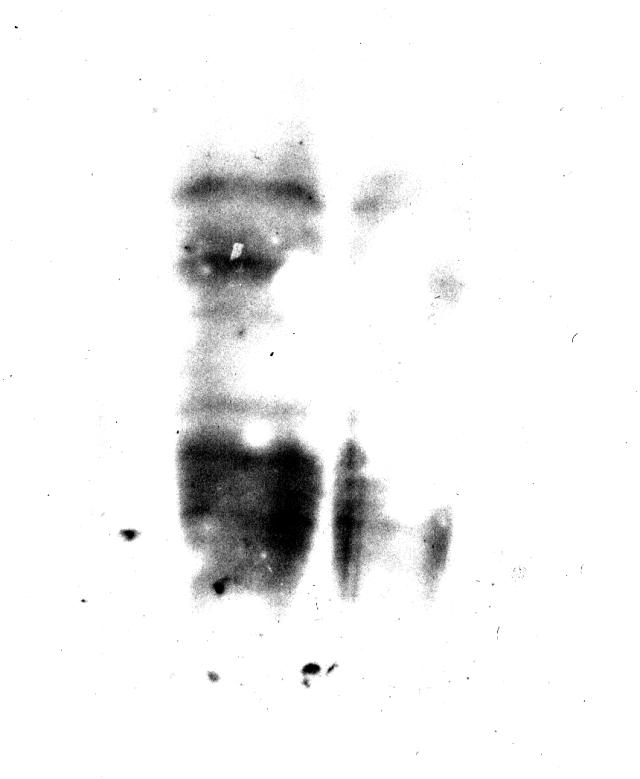 图片附件: 65309062.snap.jpg (2013-9-10 17:33, 14.87 KB) / 该附件被下载次数 12
图片附件: 65309062.snap.jpg (2013-9-10 17:33, 14.87 KB) / 该附件被下载次数 12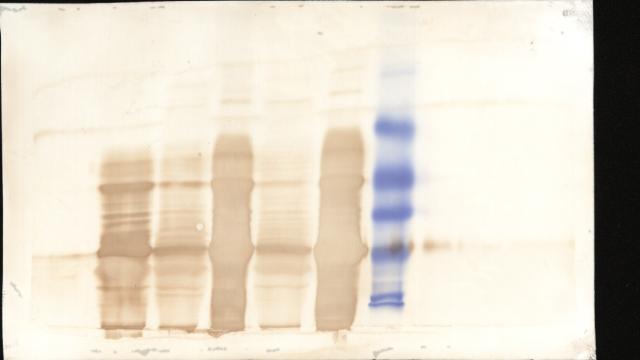 图片附件: 70186295.snap.jpg (2013-9-10 17:37, 16.28 KB) / 该附件被下载次数 75
图片附件: 70186295.snap.jpg (2013-9-10 17:37, 16.28 KB) / 该附件被下载次数 75 图片附件: 54170737.jpg (2013-9-10 18:03, 31.94 KB) / 该附件被下载次数 26
图片附件: 54170737.jpg (2013-9-10 18:03, 31.94 KB) / 该附件被下载次数 26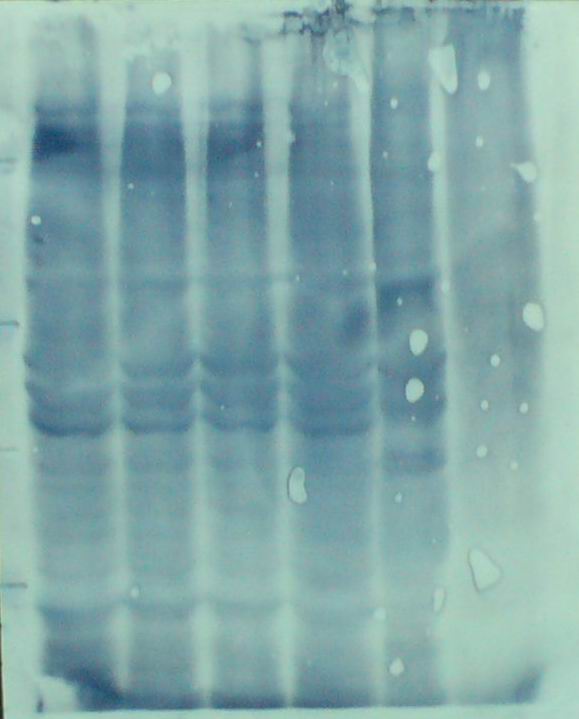 图片附件: 87218574.jpg (2013-9-10 18:07, 10.97 KB) / 该附件被下载次数 20
图片附件: 87218574.jpg (2013-9-10 18:07, 10.97 KB) / 该附件被下载次数 20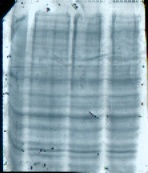 图片附件: 44452558.jpg (2013-9-11 12:57, 10.35 KB) / 该附件被下载次数 21
图片附件: 44452558.jpg (2013-9-11 12:57, 10.35 KB) / 该附件被下载次数 21 图片附件: 69096123.snap.jpg (2013-9-11 13:06, 11.15 KB) / 该附件被下载次数 17
图片附件: 69096123.snap.jpg (2013-9-11 13:06, 11.15 KB) / 该附件被下载次数 17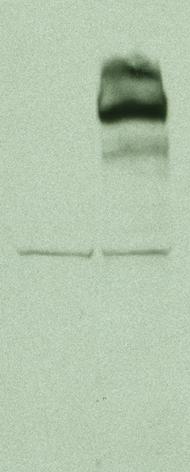 图片附件: 52914396.jpg (2013-9-11 13:24, 3.07 KB) / 该附件被下载次数 27
图片附件: 52914396.jpg (2013-9-11 13:24, 3.07 KB) / 该附件被下载次数 27 图片附件: 26899511.jpg (2013-9-11 13:58, 40.38 KB) / 该附件被下载次数 13
图片附件: 26899511.jpg (2013-9-11 13:58, 40.38 KB) / 该附件被下载次数 13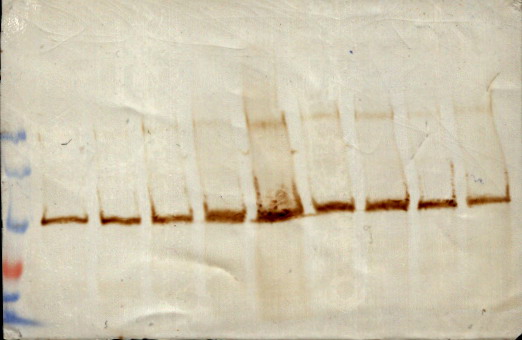 图片附件: 18708181.jpg (2013-9-11 15:16, 48.26 KB) / 该附件被下载次数 23
图片附件: 18708181.jpg (2013-9-11 15:16, 48.26 KB) / 该附件被下载次数 23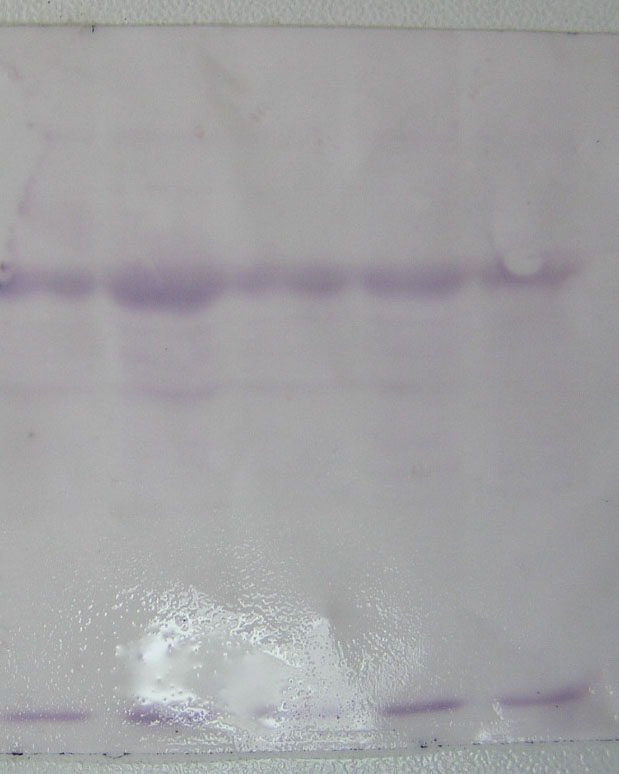 图片附件: 46859565.snap.jpg (2013-9-11 16:33, 40.85 KB) / 该附件被下载次数 21
图片附件: 46859565.snap.jpg (2013-9-11 16:33, 40.85 KB) / 该附件被下载次数 21 图片附件: 73730705.snap.jpg (2013-9-11 17:09, 32.04 KB) / 该附件被下载次数 20
图片附件: 73730705.snap.jpg (2013-9-11 17:09, 32.04 KB) / 该附件被下载次数 20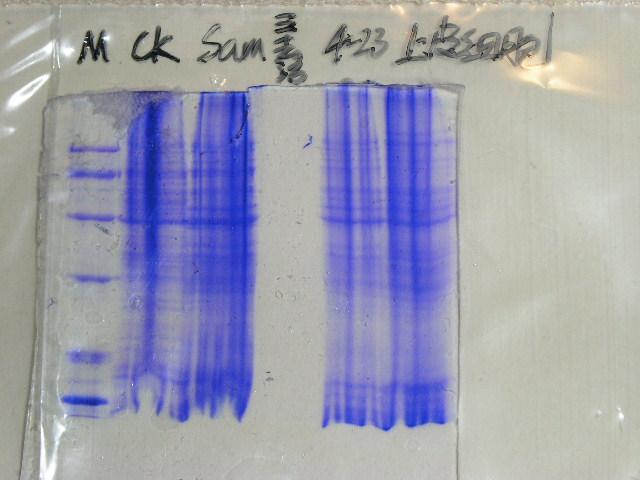 图片附件: 73861772.jpg (2013-9-11 17:11, 48.27 KB) / 该附件被下载次数 7
图片附件: 73861772.jpg (2013-9-11 17:11, 48.27 KB) / 该附件被下载次数 7 图片附件: 80787107.snap.jpg (2013-9-14 18:08, 26.57 KB) / 该附件被下载次数 14
图片附件: 80787107.snap.jpg (2013-9-14 18:08, 26.57 KB) / 该附件被下载次数 14 图片附件: 78914526.jpg (2013-9-18 13:27, 22.14 KB) / 该附件被下载次数 13
图片附件: 78914526.jpg (2013-9-18 13:27, 22.14 KB) / 该附件被下载次数 13 图片附件: 26701776.snap.jpg (2013-9-18 13:28, 20.23 KB) / 该附件被下载次数 25
图片附件: 26701776.snap.jpg (2013-9-18 13:28, 20.23 KB) / 该附件被下载次数 25 图片附件: 10720376.jpg (2013-9-18 13:39, 50.22 KB) / 该附件被下载次数 21
图片附件: 10720376.jpg (2013-9-18 13:39, 50.22 KB) / 该附件被下载次数 21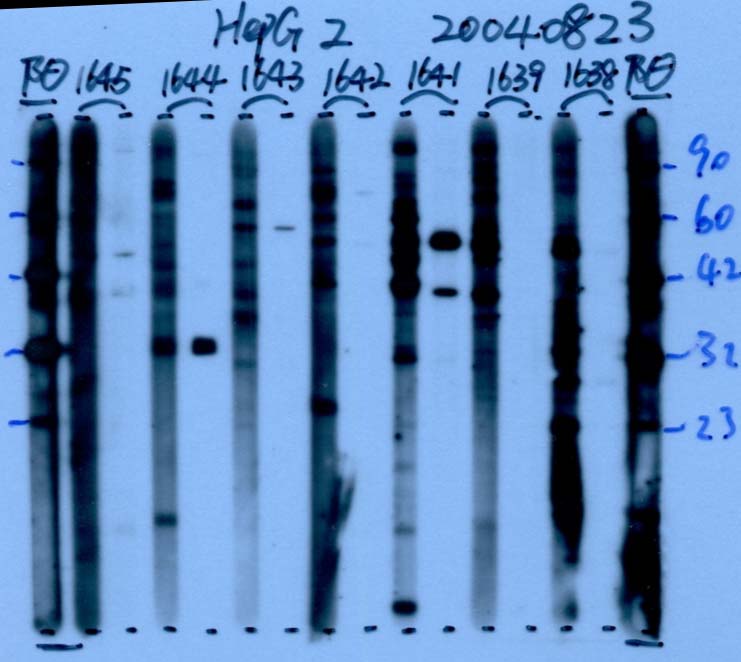 图片附件: 62931672.snap.jpg (2013-9-18 13:40, 38.63 KB) / 该附件被下载次数 23
图片附件: 62931672.snap.jpg (2013-9-18 13:40, 38.63 KB) / 该附件被下载次数 23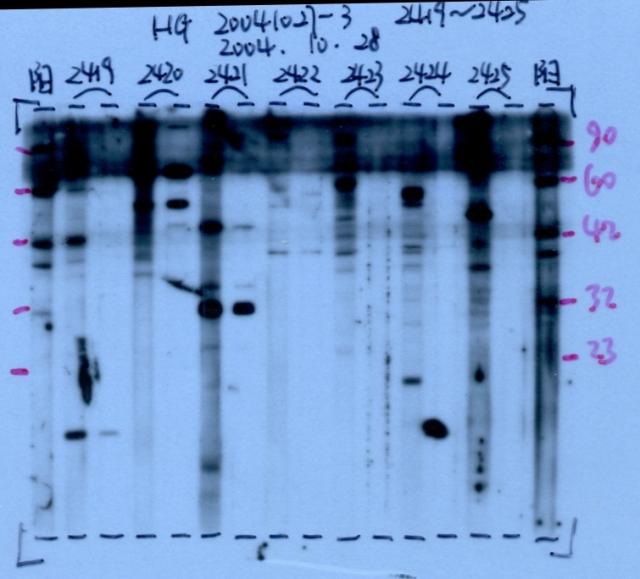 图片附件: 63685017.jpg (2013-9-18 15:11, 41.21 KB) / 该附件被下载次数 8
图片附件: 63685017.jpg (2013-9-18 15:11, 41.21 KB) / 该附件被下载次数 8 图片附件: 81314396.snap.jpg (2013-9-18 15:45, 83.88 KB) / 该附件被下载次数 23
图片附件: 81314396.snap.jpg (2013-9-18 15:45, 83.88 KB) / 该附件被下载次数 23 图片附件: 25469189.snap.jpg (2013-9-18 15:46, 61.85 KB) / 该附件被下载次数 19
图片附件: 25469189.snap.jpg (2013-9-18 15:46, 61.85 KB) / 该附件被下载次数 19 图片附件: 39003439.snap.jpg (2013-9-18 15:47, 83.88 KB) / 该附件被下载次数 20
图片附件: 39003439.snap.jpg (2013-9-18 15:47, 83.88 KB) / 该附件被下载次数 20 图片附件: 36523351.jpg (2013-9-18 16:10, 53.18 KB) / 该附件被下载次数 19
图片附件: 36523351.jpg (2013-9-18 16:10, 53.18 KB) / 该附件被下载次数 19 图片附件: 43881210.jpg (2013-9-21 13:40, 10.4 KB) / 该附件被下载次数 19
图片附件: 43881210.jpg (2013-9-21 13:40, 10.4 KB) / 该附件被下载次数 19 图片附件: 17542385.jpg (2013-9-21 13:41, 6.96 KB) / 该附件被下载次数 136
图片附件: 17542385.jpg (2013-9-21 13:41, 6.96 KB) / 该附件被下载次数 136 图片附件: 63445463.snap.jpg (2013-9-21 13:41, 4.94 KB) / 该附件被下载次数 20
图片附件: 63445463.snap.jpg (2013-9-21 13:41, 4.94 KB) / 该附件被下载次数 20 图片附件: 30949923.snap.jpg (2013-9-21 13:42, 6.84 KB) / 该附件被下载次数 22
图片附件: 30949923.snap.jpg (2013-9-21 13:42, 6.84 KB) / 该附件被下载次数 22 图片附件: 60414393.jpg (2013-9-21 14:08, 6.8 KB) / 该附件被下载次数 6
图片附件: 60414393.jpg (2013-9-21 14:08, 6.8 KB) / 该附件被下载次数 6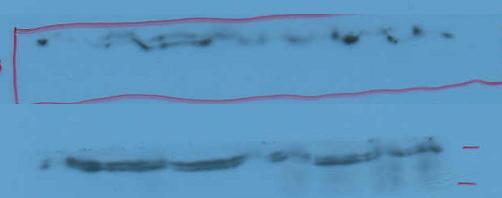 图片附件: 84330146.jpg (2013-9-22 12:24, 49.82 KB) / 该附件被下载次数 6
图片附件: 84330146.jpg (2013-9-22 12:24, 49.82 KB) / 该附件被下载次数 6 图片附件: 21469157.jpg (2013-9-22 13:24, 27.88 KB) / 该附件被下载次数 13
图片附件: 21469157.jpg (2013-9-22 13:24, 27.88 KB) / 该附件被下载次数 13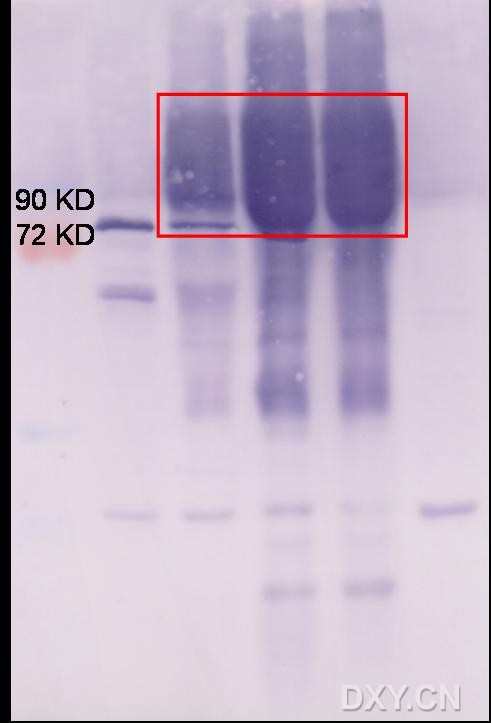 图片附件: 79386858.jpg (2013-9-22 13:29, 23.03 KB) / 该附件被下载次数 19
图片附件: 79386858.jpg (2013-9-22 13:29, 23.03 KB) / 该附件被下载次数 19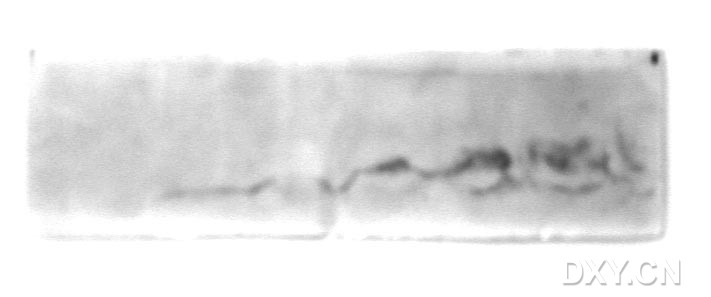 图片附件: 22377235.jpg (2013-9-22 15:25, 59.2 KB) / 该附件被下载次数 12
图片附件: 22377235.jpg (2013-9-22 15:25, 59.2 KB) / 该附件被下载次数 12 图片附件: 55596334.snap.jpg (2013-9-22 15:53, 15.07 KB) / 该附件被下载次数 3
图片附件: 55596334.snap.jpg (2013-9-22 15:53, 15.07 KB) / 该附件被下载次数 3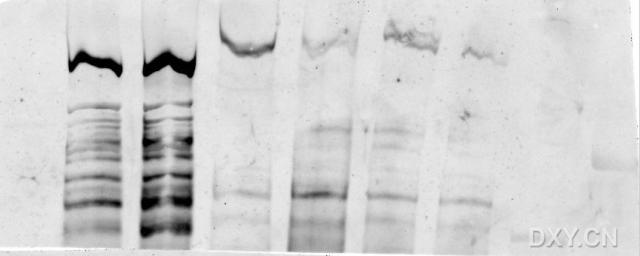 图片附件: 48814706.jpg (2013-9-22 16:29, 52.61 KB) / 该附件被下载次数 5
图片附件: 48814706.jpg (2013-9-22 16:29, 52.61 KB) / 该附件被下载次数 5 图片附件: 50836149.jpg (2013-9-22 16:40, 7.21 KB) / 该附件被下载次数 20
图片附件: 50836149.jpg (2013-9-22 16:40, 7.21 KB) / 该附件被下载次数 20 图片附件: 91361779.jpg (2013-9-22 16:50, 13.89 KB) / 该附件被下载次数 5
图片附件: 91361779.jpg (2013-9-22 16:50, 13.89 KB) / 该附件被下载次数 5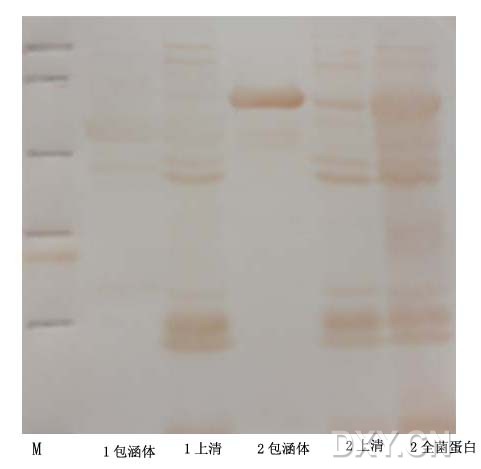 图片附件: 75536351.snap.jpg (2013-9-22 17:01, 18.08 KB) / 该附件被下载次数 18
图片附件: 75536351.snap.jpg (2013-9-22 17:01, 18.08 KB) / 该附件被下载次数 18 图片附件: 89175964.snap.jpg (2013-9-22 17:25, 9.13 KB) / 该附件被下载次数 15
图片附件: 89175964.snap.jpg (2013-9-22 17:25, 9.13 KB) / 该附件被下载次数 15 图片附件: 41621822.jpg (2013-9-22 17:28, 55.84 KB) / 该附件被下载次数 16
图片附件: 41621822.jpg (2013-9-22 17:28, 55.84 KB) / 该附件被下载次数 16 图片附件: 75749790.jpg (2013-9-22 18:14, 13.61 KB) / 该附件被下载次数 19
图片附件: 75749790.jpg (2013-9-22 18:14, 13.61 KB) / 该附件被下载次数 19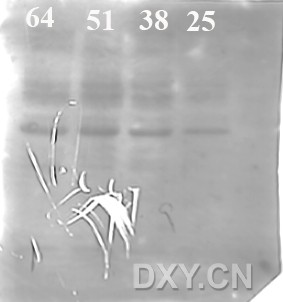 图片附件: 63333869.jpg (2013-9-22 20:06, 28.94 KB) / 该附件被下载次数 20
图片附件: 63333869.jpg (2013-9-22 20:06, 28.94 KB) / 该附件被下载次数 20 图片附件: 94852816.snap.jpg (2013-9-22 20:37, 7.77 KB) / 该附件被下载次数 25
图片附件: 94852816.snap.jpg (2013-9-22 20:37, 7.77 KB) / 该附件被下载次数 25 图片附件: 53431762.gif (2013-9-22 20:42, 28.85 KB) / 该附件被下载次数 19
图片附件: 53431762.gif (2013-9-22 20:42, 28.85 KB) / 该附件被下载次数 19
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |