标题:
【讨论帖】成功过的战友讨论下,你曾经WESTERN...
[打印本页]
作者:
nut6694
时间:
2013-10-17 17:43
标题:
【讨论帖】成功过的战友讨论下,你曾经WESTERN...
成功过的战友讨论下,你曾经WESTERN做不出来是那部分出问题了.看看各部分出问题的几率,为还没有成功,仍然苦苦寻找结果的战友提供点参考(由于本人经验有限,不足之处还请斑竹和各位大侠改正):
1、蛋白提取问题,蛋白降解,或者上样量少导致检测失败。
2、蛋白电泳失败。
3、转膜问题:转膜不充分,过转,温度过高导致蛋白结构改变等。
4、抗体问题。
5、显影问题。
6、其它。
作者:
loli
时间:
2013-10-17 17:43
抗体问题排在第一,抗体的特异性,一抗二抗的比例,这些都需要摸索条件。
蛋白电泳第二,胶的配置,由于APS的失效,tris-Hcl PH不准,丙烯酰胺的氧化(以上问题一般都是配置时间较久以后)导致电泳失败或泳道奇形怪状。
作者:
uuooii
时间:
2013-10-17 17:43
我认为western bloting 中蛋白提取是关键,最好用试剂盒提取,尤其目的蛋白是在亚细胞器表达的蛋白;其次是一抗二抗,一抗最好单克隆抗体,二抗国产好厂家的也行 ;ECL发光试剂一定要灵敏;制胶,电泳和转膜个实验室都做得比较常规了;显影定影按说明书作做活实验是经验就行。
作者:
89tongzijun
时间:
2013-10-17 17:44
我做了很久的WB,最大的一个难点感觉在于样品制备上!蛋白的降解有些时候是你想象不到的,可能出奇的糟糕,也可能完全没事~~如果单独做WB的话,感觉以“快速彻底裂解,先变性后保存”的原则比较有效,尽量选择足够强的裂解液,甚至直接给loading buffer裂解,然后取出部分蛋白定量,其余加入loading buffer100度充分变性,再分装保存。有些时候比蛋白酶抑制剂更有效。
另一个感觉就是样品中目的蛋白的量,限于WB的检测下限问题,一般目的蛋白量最好别低于1ng(虽然ECL法下限是0.1ng,即100pg),最好在10ng以上为好。这就要求在做WB之前必须充分考虑目的蛋白的可能丰度,最好充分的准备工作然后再设计细胞量或者组织量,千万别把样品搞稀了,否则就不好办了。
最后,还是多看文献,在做某个蛋白之前最好看看别人是如何做的,有什么特殊要求,内参如何选等等。
作者:
zranqi_1
时间:
2013-10-17 17:44
我以前做的时候问题主要出现在样品的制备上,也就是如果免疫时用到的蛋白和wb时的蛋白有出入,这样就会导致wb失败,所以后来我就一次性制备足够的蛋白,可以不是很纯,但是一定要和免疫动物时候用的蛋白是同一批的。
作者:
summerxx
时间:
2013-10-17 17:45
看到好几位战友都对蛋白制备有深刻的印象,也提醒我们正在WESTERN和准备WESTERN的战友,一定要注意过第一关,提出高质量的蛋白对实验成功至关重要!
希望更多的战友参与!
作者:
ha111
时间:
2013-10-17 17:45
楼上几位战友提出的蛋白制备确实非常重要,但如果在裂解液中加入cocktail的话应该不容易降解,-80度保存几个月都没有问题。
我认为western blotting 中最关键的一步是转膜。我的经验是小分子量的蛋白半干转效果比较好,大分子量(100KD)以上建议湿转。具体转膜条件需要多摸一下。30-80KD半干转恒流1mA/cm2,1-1.5h 都可以,大分子湿转100V,2.5h以上应该可以,转完膜丽春红染出的条带比较清楚的话,一般都能有结果。
抗体一般在其他问题排除后再考虑是否有质量问题。
作者:
moonlight45
时间:
2013-10-17 17:46
请教一下各位高手,我做WB时,总是内参能出来(有时条带不太完整),目的蛋白老是出不来可能是什么地方出问题?(蛋白应该没问题,同样的蛋白已经做出来几个指标了。)
还有我转膜时一次转好几张,有的出来,有的出不来可能是什么原因?
谢谢了!
作者:
nut6694
时间:
2013-10-17 17:46
请教一下各位高手,我做WB时,总是内参能出来(有时条带不太完整),目的蛋白老是出不来可能是什么地方出问题?(蛋白应该没问题,同样的蛋白已经做出来几个指标了。)
还有我转膜时一次转好几张,有的出来,有的出不来可能是什么原因?
谢谢了!
===========================================================================================================
战友,内参一般比较好做,能出来说明整个WB的流程应该没有问题,提的蛋白也可以,但是目的蛋白由于分子量大小不一样可能电泳和转膜条件也不一样,还有蛋白丰度问题导致蛋白量低出检测范围、抗体等环节都应该考虑。具体的问题可以把你详细的条件另辟新贴求助,祝出结果!谢谢!
作者:
mimili_901
时间:
2013-10-17 17:46
细节地方一定要多注意,比如SDS-PAGE时的缓冲液pH值啊,temed、ap的新鲜程度啊,样品上样前的处理啊,还有转膜时的电压/电流、膜的处理,还有ECL时的抗体孵育时的时间与温度,显影/定影啊等等。其实一般出问题很多时候都是由于在细节地方的疏忽造成的。
希望高手继续补充细节内容:)
作者:
am10
时间:
2013-10-17 17:47
我就跑胶和转膜谈一点儿自己的体会
1、跑胶 电泳Buffer重复利用的时候要注意,不能混浊,有时候能用个3-4次,有时候第二次都不行了,事先前预电泳一下,看看气泡的均匀度;Buffer一定要淹过梳子孔,否则就会发现今天的蛋白不会跑;跑的时候先用低电压跑过浓缩胶,再换高电压跑分离胶,不要追求速度,慢慢跑条带会分离的更好,特别是对于高分子量的目的蛋白;
2、转膜 我用的是湿转,Buffer一定要预冷,最好提前一天配好放4度;滤纸不要大过PVDF膜,防止短路;胶在负极,膜靠近正极,放入Tank前检查一遍会使你免以体会放反的痛心疾首;膜要标记好正反面;转膜过程中,经常过去看看,防止意外,如电泳仪接触不好。
就这些了,请各位指正
作者:
windy+++
时间:
2013-10-17 17:47
我就跑胶和转膜谈一点儿自己的体会
1、跑胶 电泳Buffer重复利用的时候要注意,不能混浊,有时候能用个3-4次,有时候第二次都不行了,事先前预电泳一下,看看气泡的均匀度;Buffer一定要淹过梳子孔,否则就会发现今天的蛋白不会跑;跑的时候先用低电压跑过浓缩胶,再换高电压跑分离胶,不要追求速度,慢慢跑条带会分离的更好,特别是对于高分子量的目的蛋白;
2、转膜 我用的是湿转,Buffer一定要预冷,最好提前一天配好放4度;滤纸不要大过PVDF膜,防止短路;胶在负极,膜靠近正极,放入Tank前检查一遍会使你免以体会放反的痛心疾首;膜要标记好正反面;转膜过程中,经常过去看看,防止意外,如电泳仪接触不好。
就这些了,请各位指正
================================================================================
我的感觉是电泳跑胶时buffer的利用,如果蛋白跑出胶外,那下次一定就要更换,如果没有的话还能用一两次.
作者:
zranqi_1
时间:
2013-10-17 17:48
我是第一次作,作了3次,目前还没有成功。我发现跑胶的时候,有时候Marker会歪歪扭扭,转完膜(90V,4hr)后还有胶孔附近有残留,是不是没有转完全?另外,DAB染色和ECL那个好一点?如何操作?
盼各位大侠给个意见。
作者:
nut6694
时间:
2013-10-17 17:48
首先确定你的胶的浓度和离子强度没问题,如果胶不均匀Marker容易跑歪。转膜后看和你的蛋白分子量近似的预染Marker有没有转过去,如果marker没问题,那基本可以认为你的蛋白转过去了。显色的话应该ECL好些吧,灵敏度更高呢,HRP标记的二抗孵育完后用TBST充分清洗,然后加ECL试剂,包好后压片,一般如果你能够看到荧光的话,压片5min以内就可以看一下,如果看不见的话,直接压半小时吧。暗室仲压片,压片完后显影、定影、水洗,就可以了。
作者:
xue258
时间:
2013-10-17 17:49
做了几个月western了,总结下本人在western中遇见的一些问题:
听过一个讲座,说是在抗体中加叠氮钠,可以在4度保存,避免抗体的反复冻融。结果我就犯错误了,二抗也加叠氮钠了,导致发光显不出条带,找原因找了一周,才知道叠氮钠能抑制HRP的活性。
作者:
S6044
时间:
2013-10-17 17:49
我所纯化的蛋白为未知蛋白,目前仅知其具有甘露糖结合的活性和分子量,想用WB的方法鉴定纯化出的蛋白是否具有结合甘露糖的能力。在实验的过程中一直得不到理想的结果,有两个问题请各位高手指教:
1、不知在SDS-PAGE和WB的条件下,蛋白的糖结合活性是否可得以保存
2、因为蛋白是未知的,所以没有抗体可用,我们用一种带有甘露糖基的BSA来代替抗体,并用10nm胶体金与之耦联作为显色之用。10nm胶体金之前一直用于电镜观察,不知可否用于WB的显色。请各位各个意见。
作者:
JK.jon
时间:
2013-10-17 17:49
我作WB,
第一次:SDS-PAGE电泳都没跑出来,怀疑是蛋白降解了,但是后来同样的样品和别人一起又跑一次,就跑出来了,发现是配胶的问题。分离胶要用12%的。
第二次:样品制备OK,浓度也测好了,电泳也跑得不错,PVDF膜不小心被手指划了一下,结果最后显影时候,显出来一个划痕。
第三次:我的B-actin做出来了,但是我的目的条带怎么也不出来,同样的条件,同样的过程,试剂也一样,我师姐的就做出来了,可见不是试剂的问题,显影同样用的DAB,这次考虑是抗体的问题了。
第四次:找公司换了抗体,果然ok了!
作者:
eric930
时间:
2013-10-17 17:50
本底过高:
(1)封闭不好。
(2) 一抗、二抗作用强度过高。
(3) 洗涤不充分。
对策包括:
a. 延长封闭时间;
b. 可调节一抗,二抗的稀释度,找到一个最佳浓度,使之能得到有效强度的信号,而非特异性的染色较低。
c. 充分洗涤,将未结合上的非特异性的抗体洗掉,可在缓冲液中加0.02-0.2%Tween20。
假阳性: 多数由抗体和有部分同源性的抗原间的交叉反应引起。主要解决办法是在保证阳性结果可见的前提下,尽可能高地提高抗体稀释度,使假阳性消失
作者:
mamamiya
时间:
2013-10-17 17:51
我进实验室最先接触的就是WESTERN,我觉得首先要提出高质量的蛋白,然后一鼓作气往下做,不要放太久,即使-80度超过三个月结果也不太理想。新鲜的样品跑出来的条带很靓的。
实验室的温度对制胶很重要,气温过低会使凝胶不均匀,一、二抗也孵不上去,夏天的时候,不到8个小时就可以做完一个WESTERN,现在就不行了。
封闭液要保证有效。
一、二抗比例要适当,不然即使高浓度也没用。
转膜是个技术活,要耐心,但不是磨蹭。
化学发光试剂要好。
显影时间的控制和调整。
另外注意一些细小问题,比如缓冲液不要加错,上样要尽快,电泳时电压不要太高,洗膜要充分等,有时候往往会犯一些及其可笑的错误。
作者:
阿k
时间:
2013-10-17 17:51
如果胶中有小的裂隙,对转膜有影响吗?
作者:
tudou85
时间:
2013-10-17 17:51
我最近一直在做,可是碰见一下问题向高手们请教,希望提点改正意见:
1 转膜时,多次出现小分子量的转膜效果不如中分子和大分子量的(用的彩虹MARKER,但14KD的亮黄色的条带仍然在胶上,转膜后的胶染色后小分子量区域的蛋白残留较多。在整张和将不同区域的分别转膜时均出现这种情况。)
2 在暗室里肉眼可以看见发光,可是胶片上一无所有。
3 ECL没有表达,可是用DAB时膜上却出现条带。
4 手工显影时,胶片的显影时间和温度有很大关系吗?显影的时间掌握如何比较恰当?
作者:
mimili_901
时间:
2013-10-17 17:52
我最近做WB,为了更进一步证实我的目的蛋白,我加了标准品,但也许是我的目的蛋白表达量太少,所以曝光总是很弱,甚至没有,但每回都能将标准蛋白曝光到片子上。另我感到奇怪 的是,每次我曝光后再将膜用丽春红染色,标准蛋白在膜上几乎看不见,但对应的位置却可以看见明显的条带。所以我想问一下各位高手,是否我看到与标准蛋白对应位置的明显条带其实是很多种蛋白的混合物,也许没有我所要的目的蛋白,或者即使有,也是非常少,少得不足以可以曝光在片子上。还有一个问题,如果目的蛋白表达很好的话,加大抗体浓度有用吗?实验很着急,希望尽快回复。多谢了!!!
作者:
nut6694
时间:
2013-10-17 17:52
新话题:考染胶有目的条带、丽春红染膜有条带和压片有条带这三者之间有什么必然的联系吗?请根据大家的经验谈谈。
作者:
kuaizige
时间:
2013-10-17 17:52
QUOTE:
原帖由
nut6694
于 2013-10-17 17:52 发表
bbcodeurl('http://bbs.antpedia.com/images/common/back.gif', '
%s
')
新话题:考染胶有目的条带、丽春红染膜有条带和压片有条带这三者之间有什么必然的联系吗?请根据大家的经验谈谈。
考染胶有目的条带:说明你的样品中有你要检测的蛋白。
丽春红染膜有条带:说明你的目的蛋白已经成功转移到膜上(有没有完全就不一定,可能也没有必要知道,一定要检查,或者丽春红没有条带,可以将电转后的胶再考染)。
压片有条带:目的蛋白与抗体有特异性反应。
由于检测限是western>考染>丽春红,所以丽春红有条带则考染,压片也应该有,反之则不一定。
再说说我的一些体会。
电转开始,有许多战友会提到短路问题,我觉得从理论到实践都说不通。首先,整块胶都被导电的buffer包围,何来通路、短路?其次,我,包括我们实验室其他人,从来没有按胶的尺寸来剪膜和滤纸。我的做法是剪一块比胶略大的膜(不便宜,省着点用),滤纸则可以同夹板一样大小,干的、湿的从来没有出现过问题。
气泡是最主要的问题,最好将夹胶、膜的操作在buffer液面下进行。
电转buffer最好不要太小气,用3-4次就该换了,反正也不贵,配也简单。
丽春红没有必要每次用,一般电转没有问题。
一抗要是血清的话,可以四℃过夜。
二抗按说明书来基本没有问题。
ECL显色后,尽量将显色液除尽,可以将膜封入一次性手套(封三边,留一边),用吸水纸使劲擦,再封好压片。
要是对曝光时间没有把握,可以一次叠两张胶片,相当于做个梯度。
不当之处望各位指正。
作者:
youyou99
时间:
2013-10-17 17:53
求教各位高手,最近我的Western blot经常是出来干干净净,什么都没有,成功率低。经过SDS-PAGE胶考马斯亮蓝染色,证明电泳没有问题,蛋白也没有降解。用正对照蛋白做点杂交也证明一抗,二抗以及发色系统没有问题。
疑点就集中在转膜过程上,因为之前试验正对照蛋白(30KD)的时候用的转膜用的厚吸水纸用完了(“三明治”两侧各一张),换成了薄的滤纸(两侧各三张),自从换了这个以后就一直有问题了,即使正对照也很难做出来。我用的是湿转法,转膜buffer同《分子克隆》上:48mM Tris,39mM 甘氨酸,20%甲醇,0.0375%SDS,转膜条件:4度,200mA稳流一小时,我的蛋白比较大,73KD,这个条件是否合适?转膜结束之后将胶用考马斯亮蓝染色,干干净净,说明已经转走了,将PVDF膜用丽春红染色,也什么也看不到,感觉不太可能转过头,因为之前30KD的蛋白用这个条件也可以的。最近我又将转膜buffer的甲醇比例下调到15%,其他条件相同。结果也是一样失望,各位有什么建议么?
作者:
+小生怕怕+
时间:
2013-10-17 17:53
我做了很久的WB,最大的一个难点感觉在于样品制备上!蛋白的降解有些时候是你想象不到的,可能出奇的糟糕,也可能完全没事~~如果单独做WB的话,感觉以“快速彻底裂解,先变性后保存”的原则比较有效,尽量选择足够强的裂解液,甚至直接给loading buffer裂解,然后取出部分蛋白定量,其余加入loading buffer100度充分变性,再分装保存。有些时候比蛋白酶抑制剂更有效。
另一个感觉就是样品中目的蛋白的量,限于WB的检测下限问题,一般目的蛋白量最好别低于1ng(虽然ECL法下限是0.1ng,即100pg),最好在10ng以上为好。这就要求在做WB之前必须充分考虑目的蛋白的可能丰度,最好充分的准备工作然后再设计细胞量或者组织量,千万别把样品搞稀了,否则就不好办了。
-------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
师兄或师姐,你说的意思是直接把提取的蛋白液加入loading buffer煮沸变性后直接保存,然后蛋白电泳时直接拿出来上样就可以吗?那样保存可以吗?加入loading buffer 变性后的蛋白样放几度保存?四度就行还时-20或时-80度?
第二,你说的是加样量少可能检测不到,那么如果加样量非常多,对实验是不是也会又很大影响?
作者:
+小生怕怕+
时间:
2013-10-17 17:54
我就跑胶和转膜谈一点儿自己的体会
1、跑胶 电泳Buffer重复利用的时候要注意,不能混浊,有时候能用个3-4次,有时候第二次都不行了,事先前预电泳一下,看看气泡的均匀度;Buffer一定要淹过梳子孔,否则就会发现今天的蛋白不会跑;跑的时候先用低电压跑过浓缩胶,再换高电压跑分离胶,不要追求速度,慢慢跑条带会分离的更好,特别是对于高分子量的目的蛋白;
2、转膜 我用的是湿转,Buffer一定要预冷,最好提前一天配好放4度;滤纸不要大过PVDF膜,防止短路;胶在负极,膜靠近正极,放入Tank前检查一遍会使你免以体会放反的痛心疾首;膜要标记好正反面;转膜过程中,经常过去看看,防止意外,如电泳仪接触不好。
就这些了,请各位指正
======================================================================
滤纸不要大过PVDF膜??哎,,到底滤纸,膜.胶的大小顺序是什么啊 怎么每个人说法都不一样啊
作者:
+小生怕怕+
时间:
2013-10-17 17:54
谢谢战友!我的感觉是电泳跑胶时buffer的利用,如果蛋白跑出胶外,那下次一定就要更换,如果没有的话还能用一两次.
=============================================================================================
什么叫蛋白跑出胶外呢?我每次都把兰色那条带跑出胶,蓝色那条跑到快底部,那算是蛋白跑出胶外吗?我的蛋白样是组织提取液
作者:
+小生怕怕+
时间:
2013-10-17 17:55
换成了薄的滤纸(两侧各三张),自从换了这个以后就一直有问题了,正对照也很难做出来。
==============================
滤纸用的张数对转膜有很大的影响吗?
作者:
S6044
时间:
2013-10-17 17:55
湿法转移转移的确从来没什么短路的情况,半干转移才要特别注意,不过后者基本上不用,我们做了比较的,稳定性和重复性前者要好得多,当然对于高手来说也无所谓,但我觉得能做湿法的尽量还是用湿法。
1. 这种情况下我们通常会同时跑两块胶(或者一块胶切成两半),一块染色一块做Western,同步试验才能确保电泳没有问题,不知道你是不是这样做的。
2. 转膜我们一直用的25mM tris 192mM Glycine 15%甲醇(两次一换),各个实验室会用一套比较成熟的系统,如果你们的系统一直很好我觉得你的buffer你也不用怀疑它,只不过要注意check一下你的PH值。一旦摸索出较好的系统后不要轻易更换所使用的试剂厂家。
3. “考马斯亮蓝染色,干干净净,PVDF膜用丽春红染色,也什么也看不到”
的确,73KD这种条件下不会转过,所以我觉得是不是你的电极插线或者是做三明治的夹具放反了,全部转到缓冲液中去了,滤纸不会有什么问题,两种都可以,我们在没有厚的专用滤纸前一直用的是3张普通滤纸。
4. 没有用过的抗体最好有阳性对照,甚至我们每次做都会使用阳性对照,确保我们在出了问题能迅速找到原因
如果手头没有什么成熟的western体系可以向各个做抗体的大公司索要,这些体系都非常常熟,自己练习两次就熟悉了。
作者:
BOSS2011
时间:
2013-10-17 17:55
我这几个月也做了十多次WB, 最大的感触就是提取高质量的蛋白很关键。
作者:
wu11998866
时间:
2013-10-17 17:56
相关疾病:
头痛
我最近也一直在做小歪,感觉成功的关键在于封闭之前的步骤,毕竟我们的抗体大都购与大公司,不会有什么问题,定夺也就做个dot blotting摸个比例,
但是蛋白制备,sds page,转膜我认为与个人操作有关,我的同界同学竟然把marker转到膜上后,发现目的蛋白还在胶上(不要怀疑,真的是这样,就是不知道为什么).
还有就是蛋白的位置,我要检测260k的一个蛋白,这个实验目前还没有成功,因为我总是没有办法让它跑到理想的位置.头疼啊
蛋白制备没有什么好说的了,好多方法,不停的实验找到合适自己的就ok
作者:
tie8
时间:
2013-10-17 17:56
滤纸用的张数对转膜有很大的影响吗?
=================================
我也觉得没有影响,但现在就是非常奇怪。
作者:
tie8
时间:
2013-10-17 17:57
湿法转移转移的确从来没什么短路的情况,半干转移才要特别注意,不过后者基本上不用,我们做了比较的,稳定性和重复性前者要好得多,当然对于高手来说也无所谓,但我觉得能做湿法的尽量还是用湿法。
===========================================================================================================
1. 这种情况下我们通常会同时跑两块胶(或者一块胶切成两半),一块染色一块做Western,同步试验才能确保电泳没有问题,不知道你是不是这样做的。
2. 转膜我们一直用的25mM tris 192mM Glycine 15%甲醇(两次一换),各个实验室会用一套比较成熟的系统,如果你们的系统一直很好我觉得你的buffer你也不用怀疑它,只不过要注意check一下你的PH值。一旦摸索出较好的系统后不要轻易更换所使用的试剂厂家。
3. “考马斯亮蓝染色,干干净净,PVDF膜用丽春红染色,也什么也看不到”
的确,73KD这种条件下不会转过,所以我觉得是不是你的电极插线或者是做三明治的夹具放反了,全部转到缓冲液中去了,滤纸不会有什么问题,两种都可以,我们在没有厚的专用滤纸前一直用的是3张普通滤纸。
4. 没有用过的抗体最好有阳性对照,甚至我们每次做都会使用阳性对照,确保我们在出了问题能迅速找到原因
如果手头没有什么成熟的western体系可以向各个做抗体的大公司索要,这些体系都非常常熟,自己练习两次就熟悉了。
1.每次我都是跑一份平行的SDS-PAGE考染的,一切正常。
2.你所说的buffer是半干法的buffer吧,这套湿转膜系统以前本实验室一直用的。
3.这个电极搞反或是三明治搞错是不可能的,我做过n次,以前都是好的,突然就不行了,一直到现在,我每次都仔细检查,若是偶尔一次还有可能,连续多次就不可能了。
4.我前面说了,正对照是有的,点杂交出来,就是western出不来。
作者:
tie8
时间:
2013-10-17 17:57
湿法转移转移的确从来没什么短路的情况,半干转移才要特别注意,不过后者基本上不用,我们做了比较的,稳定性和重复性前者要好得多,当然对于高手来说也无所谓,但我觉得能做湿法的尽量还是用湿法。
1. 这种情况下我们通常会同时跑两块胶(或者一块胶切成两半),一块染色一块做Western,同步试验才能确保电泳没有问题,不知道你是不是这样做的。
2. 转膜我们一直用的25mM tris 192mM Glycine 15%甲醇(两次一换),各个实验室会用一套比较成熟的系统,如果你们的系统一直很好我觉得你的buffer你也不用怀疑它,只不过要注意check一下你的PH值。一旦摸索出较好的系统后不要轻易更换所使用的试剂厂家。
3. “考马斯亮蓝染色,干干净净,PVDF膜用丽春红染色,也什么也看不到”
的确,73KD这种条件下不会转过,所以我觉得是不是你的电极插线或者是做三明治的夹具放反了,全部转到缓冲液中去了,滤纸不会有什么问题,两种都可以,我们在没有厚的专用滤纸前一直用的是3张普通滤纸。
4. 没有用过的抗体最好有阳性对照,甚至我们每次做都会使用阳性对照,确保我们在出了问题能迅速找到原因
如果手头没有什么成熟的western体系可以向各个做抗体的大公司索要,这些体系都非常常熟,自己练习两次就熟悉了。
==========================================================================================================
1.每次我都是跑一份平行的SDS-PAGE考染的,一切正常。
2.你所说的buffer是半干法的buffer吧,这套湿转膜系统以前本实验室一直用的。
3.这个电极搞反或是三明治搞错是不可能的,我做过n次,以前都是好的,突然就不行了,一直到现在,我每次都仔细检查,若是偶尔一次还有可能,连续多次就不可能了。
4.我前面说了,正对照是有的,点杂交出来,就是western出不来。
作者:
greenbee
时间:
2013-10-17 17:58
我做western开始条带不出来,但帮别人做的全部出来了,后来发现是一抗坏了,好容易说动老板重新买一抗,发现不能太帮老板省钱,要不辛苦倒霉的还是自己。
后来做stripper,开始的时候都能出来,后来一次偷懒,自己没配用了别人配的,结果就做不出来了,发现做实验一点不能马虎,东西最好全部自己配置,还要及时换新的。
western做多了,几个同时做,还出现过拿错二抗的事情,呵呵,当时就纳闷怎么很熟练的条件突然一点条带也没有了。
作者:
yayya
时间:
2013-10-17 17:59
但是蛋白制备,sds page,转膜我认为与个人操作有关,我的同界同学竟然把marker转到膜上后,发现目的蛋白还在胶上(不要怀疑,真的是这样,就是不知道为什么).
===========================================================================================================
你说marker 转到膜上了 目的蛋白还在胶上 这样的情况很正常啊
我们实验室很多师姐就遇到过这样的情况 她们做的蛋白大的时候好象尤其这样 最近我也有过这样的情况 做42KD的蛋白还总是偶尔压片出来带时 转膜都是marker已经转没了
到是marker清晰可见在膜上时 多次还压不出带呢
作者:
yayya
时间:
2013-10-17 17:59
我这几个月也做了十多次WB, 最大的感触就是提取高质量的蛋白很关键。
====================================================
那么你现在是否有提取高蛋白质量的方法呢? 分享一下吧
我看很多人似乎都用组织匀浆器来提取组织总蛋白
比如匀三秒停三秒 共匀15秒 什么的
有的又提倡最好别用组织匀浆器 因为产热大 而且不易控制不起沫
我们实验室一直用玻璃匀浆器 迅速冰上进行
但是我自己做起来 总觉得"迅速"这两字做起来好难啊
尤其在组织剪碎不完全的情况下 最后发现组织块根本就匀不碎了
所以最后我得出的教训就是:一定要在匀浆之前把组织块充分匀碎
作者:
yayya
时间:
2013-10-17 17:59
我做western开始条带不出来,但帮别人做的全部出来了,后来发现是一抗坏了,好容易说动老板重新买一抗,发现不能太帮老板省钱,要不辛苦倒霉的还是自己。
后来做stripper,开始的时候都能出来,后来一次偷懒,自己没配用了别人配的,结果就做不出来了,发现做实验一点不能马虎,东西最好全部自己配置,还要及时换新的。
western做多了,几个同时做,还出现过拿错二抗的事情,呵呵,当时就纳闷怎么很熟练的条件突然一点条带也没有了。
==============================
想问你 你stripper 用的溶液与步骤?
谢谢
作者:
moonlight45
时间:
2013-10-17 18:00
个人感觉做wb首要的一步也是最关键的一步就是提蛋白,相对于细胞蛋白来说,组织的蛋白更难提取一些(蛋白的纯度和降解方面不易控制),上面的战友也提到了组织匀浆器和玻璃匀浆器迅速冰上进行的方法,个人感觉为避免蛋白的降解最好采用液氮研磨的方法,组织块不用剪的太碎也可以,研磨过程中注意补充液氮就可以了,不受时间的限制,唯一的缺点就是对组织有一定的损失。
作者:
DONT
时间:
2013-10-17 18:00
做了十多次WB,第一次可笑的错误是,将10*转移缓冲液配成1*时忘了加甲醇(配方是在稀释时加甲醇的)。还有一次是错将1:1000抗体配成1:10000。还有就是将用于化学发光的建议抗体稀释度来做DAB显色,结果什么也没有,用化学发光还是可以做出来的。
作者:
04906
时间:
2013-10-21 14:59
我的蛋白样品浓度为1.2mg/ml,样品上样量为20,样品是制备好直接上样,可是显色后目标条带非常浅,后来用新的一抗、二抗结果一样。可是以前几次蛋白浓度只有0.2mg/ml时,目标条带都非常清晰,请各位高手指教。
作者:
98776langtao
时间:
2013-10-21 14:59
还有问一下转膜条件的问题,看到很多资料上用的是恒流200mA转膜,有的资料上则是恒压60V转膜,恒流和恒压有什么区别么?
作者:
greenbee
时间:
2013-10-21 15:02
请问,用正对照蛋白做点杂交也证明一抗,二抗以及发色系统没有问题,具体是怎么做的啊?我现在想证明是一抗还是二抗出现问题,谢谢啊!
作者:
plaa
时间:
2013-10-21 15:02
直接用2抗在 纤维膜上dab或ecl显色。
结果可出来 则2抗没问题。
2抗没问题就 可以检测1抗。
作者:
plaa
时间:
2013-10-21 15:04
求教各位高手,最近我的Western blot经常是出来干干净净,什么都没有,成功率低。经过SDS-PAGE胶考马斯亮蓝染色,证明电泳没有问题,蛋白也没有降解。用正对照蛋白做点杂交也证明一抗,二抗以及发色系统没有问题。
疑点就集中在转膜过程上,因为之前试验正对照蛋白(30KD)的时候用的转膜用的厚吸水纸用完了(“三明治”两侧各一张),换成了薄的滤纸(两侧各三张),自从换了这个以后就一直有问题了,即使正对照也很难做出来。我用的是湿转法,转膜buffer同《分子克隆》上:48mM Tris,39mM 甘氨酸,20%甲醇,0.0375%SDS,转膜条件:4度,200mA稳流一小时,我的蛋白比较大,73KD,这个条件是否合适?转膜结束之后将胶用考马斯亮蓝染色,干干净净,说明已经转走了,将PVDF膜用丽春红染色,也什么也看不到,感觉不太可能转过头,因为之前30KD的蛋白用这个条件也可以的。最近我又将转膜buffer的甲醇比例下调到15%,其他条件相同。结果也是一样失望,各位有什么建议么?
=============================================
试一下这个buffer : 甘氨酸1.45 g
tris碱2.9g SDS 0.185g 甲醇100ml 加去离子水补足500ml
室温存放
作者:
lxh031
时间:
2013-10-21 15:08
做了近一年的WESTERN,刚开始一直没出来,最后改变了变性温度,由90度降至70度,目的带就出来了,所以在蛋白变性这个环节也是很重要的.
作者:
one
时间:
2013-10-21 15:08
相关疾病:
头痛
自从进实验室就一直做WB,成功失败都经历过,些许经验教训愿与大家分享。
大家都知道WB有几个关键因素,分别是:蛋白,电泳,转膜,一抗,显影(或显色)。其中最重要的我认为是一抗和蛋白问题。
1.蛋白 (1)提取时原则主张使用玻璃匀浆,因超声匀浆可使蛋白降解。但前者耗时较多,即使冰上操作亦会有降解的。实验中发现超声匀浆也不错(2)裂解液 如提取后测蛋白浓度较低可少加一点。不主张加上样缓冲液,因最好每次做之前都测一下浓度。(3) 保存:-20保存的话建议一周内使用
2.电泳 各实验室均有自己的条件,应该都可以。注意的是加样不足十孔时,两侧加同体积上样缓冲液 。加相同蛋白量体积不同时用上样缓冲液补齐,保证相同上样体积
3.转膜 相同分子量的蛋白使用过不同的条件,效果相差不多。注意偏大或偏小的蛋白条件不同即可
4.封闭 做过肾脏和心肌的背景都很深,加大封闭液浓度至8%~10%,加入1%的BSA会好一点
5.一抗 最重要!曾经因为一个指标换过所用的条件就是不出,最后发现是一抗的问题,之前一直被告知抗体很好,所以没考虑。多花点钱买个好的单克隆抗体省时省力,少头痛!
6.显影 用ECL的话,如看到膜上有条带,我们压片十几秒就可以,可以反复多压几张片子。对了,在加发光液时,要注意两侧的条带,不然两侧的条带会较弱或显不出。
以上为个人意见,仅供参考。
作者:
yysr238
时间:
2013-10-21 15:09
楼主提议不错,浏览一下,收获不小。
本人也有些经验以供分享:
楼上好多提议作WB最重要的是提高蛋白浓度,偶颇有同感,但也有例外。首先要看你实验目的,如果作2D或者低丰度蛋白就必须尽量提高蛋白浓度,一般组织可以达到30ug/ul,细胞可以到10ug/ul(本人经验)。
其次看你作的蛋白的需求量,如果仅需要10ug左右,就不要搞很高的浓度,因为定量时稀释倍数太大,结果就会很差。同时上样量太少误差就很明显。把上样体积定在10-20ul最好。
我做细胞比较多,收集了一些裂解细胞的方法,仅供参考:
1) 全细胞裂解液150mmol/L NaCl, 1mmol/L EGTA, 50mmol/L Tris-HCl(pH 7.4), 1%NP-40, 0.5μg/ml Leupeptin, 0.7μg/ml Pepstatin A, 50μg/ml PMSF, 2.2μg/ml Aprotinin。
在4℃条件下,按5×106个细胞加100ul含有蛋白酶抑制剂的细胞裂解液,蛋白酶抑制剂用前临时加入。在冰上反应30min后,4℃,12000r/min离心10min收集上清,-80℃保存备用。
2)胞浆蛋白、胞核蛋白抽提裂解液及操作步骤:
a)胞浆蛋白抽提液:10 mM HEPES (PH7.8), 15 mMKCl, 10% N P-40, 0.1mM EDTA , 1 mM DTT, 1 mM MgCl2,使用前加蛋白酶抑制剂终浓度为100μg/ml PMSF, 2.35μg/ml aprotinin, and 5μg/ml leupeptin,0.7μg/ml。4℃储存。
b)胞核裂解液:20mM HEPES (7.9),10mM KCl , 1mM EDTA,1mM EGTA,1mM DTT, 20 mM NaF, 1 mM Na4P2O7,1 mMNa3VO4, 420mM NaCl, 20%glycerol, 使用前加100μg/ml PMSF, 2.35μg/ml aprotinin, and 5μg/ml leupeptin,0.7μg/ml。4℃储存
c)蛋白抽提:细胞5x l06,用PBS(PH7.2)洗1次,以下在冰上操作。弃上清,加入100-150μl胞浆裂解液,混悬,冰上肿胀15min,再剧烈震荡涡动10s。10000g,离心20s(4o):仔细吸取全部上清,为胞浆蛋白-80o保存备用。沉淀加核蛋白裂解液50-100μl混悬,冰浴30min,间以搅拌。12000g,5min取上清分装,为胞核蛋白-80o保存备用。
作者:
yysr238
时间:
2013-10-21 15:09
转自dxy一个配方
裂解buffer:
50mM HEPES ph7.0
1mM 甘油
1mM DTT(还原剂)
7M 尿素,2M硫脲(可以提高膜蛋白的融解)
1mM EDTA(金属鏊合剂)
1mM PMSF,1mg/ml leupeptin,1mg/ml aprotinin,1mg/ml pepstatin(蛋白酶抑制剂)
50mM NaF(ph7.0)(anti hemoaggulating)
1mM Na3VO4(ph7.0)(inhibitor of phosphatases)
2mM benzamidine(inhibitor of trypsin)
作者:
9900
时间:
2013-10-21 15:09
我是新手上路,想请教一下,我的二抗是HRP标记的,是不是要用DAB显色?在那可以买到这种显色的试剂盒?请各位大虾指教.
作者:
nn255
时间:
2013-10-21 15:10
做了几个月western了,总结下本人在western中遇见的一些问题:
听过一个讲座,说是在抗体中加叠氮钠,可以在4度保存,避免抗体的反复冻融。结果我就犯错误了,二抗也加叠氮钠了,导致发光显不出条带,找原因找了一周,才知道叠氮钠能抑制HRP的活性。
------------------------------------------------------------------------------------------------------------------
我和你犯的错误一样啊!
以前做很成功,自从加了叠氮钠就连连失败,正怀疑是不是叠氮钠加的不好呢!
兄台,多谢提醒!
作者:
mysmdbl
时间:
2013-10-21 15:14
我做western blot 的一点经验
蛋白的提取方面,千万不要图省事,蛋白裂解液中该加的试剂一定要加全了,保证每次裂解的条件一样,蛋白定量后最好分装,避免反复冻融。
转膜时,气泡要赶完全,以免转膜不完全。
曝光的时候如果某个泳道的某条带残缺,出得不全,可以考虑马上再加些显色剂,作用片刻再曝光,一般问题可以解决。
有些发光剂不稳定,用到最后的时候,可以发光,但是可能淬灭的比较快,可能会影响显色结果。
作者:
mysmdbl
时间:
2013-10-21 15:15
我最近一直在做,可是碰见一下问题向高手们请教,希望提点改正意见:
1 转膜时,多次出现小分子量的转膜效果不如中分子和大分子量的(用的彩虹MARKER,但14KD的亮黄色的条带仍然在胶上,转膜后的胶染色后小分子量区域的蛋白残留较多。在整张和将不同区域的分别转膜时均出现这种情况。)
2 在暗室里肉眼可以看见发光,可是胶片上一无所有。
3 ECL没有表达,可是用DAB时膜上却出现条带。
4 手工显影时,胶片的显影时间和温度有很大关系吗?显影的时间掌握如何比较恰当?
===========================================================================================================
2 这个现象在你的目的带好像不大可能,我们倒是在使用普利来的MARKER时出现过这种问题,在暗室可以看到MARKER发光,但是胶片上什么都没有,后来发现是marker在每次使用后需要在可见光下暴露片刻才可继续使用,否则虽然在暗处发光,但是不会让胶片感光的。
3 用DAB时膜上出现条带,说明你的蛋白转过去了,而且二抗也结合上了。如果ECL没有表达,是不是你的ECL发光剂过期了或者不够敏感呢?
4温度对显影影响不大。做western 你当然要做预实验摸一下条件了,包括一抗和二抗的浓度、显色时间。显影的时候以目的带比较清楚又不能太浓为宜,如果太浓,可能会看不出差异,尤其是在对内对照显影的时候,不要太浓,曝光浅些会比较漂亮。
作者:
plaa
时间:
2013-10-21 15:15
我个人认为,Western的关键问题是抗体问题,一抗,二抗都很关键,我曾经有一个月就因为二抗出了问题而没有结果。还有抗体的条件很难摸,不同的时间段都不一样。
作者:
orangecake
时间:
2013-10-21 15:16
我前一段时间杂了很久的白板(ECL)
原因有二:
1 我使用的样品是提取好的蛋白冻在-80℃,降解了,我就设了阳性对照,阳性信号也不强。困惑!
2 我后来查出一抗都在4℃放了两年了,好像保质期是一年!!狂倒
我后来自己提蛋白,换了新抗体,万事大吉!
我认为能不能有结果抗原是关键,抗原蛋白的量太低,提高一抗浓度是解决不了问题的。
至于能不能作出漂亮的结果那就要看抗体浓度了,当然这个抗体要能用。
再有二抗时间不能太长,要不片子很丑!!!呵呵
作者:
ladyhuahua
时间:
2013-10-21 15:16
老大,不是电极方向反了吧?
作者:
yjf1026
时间:
2013-10-21 15:17
湿法转移转移的确从来没什么短路的情况,半干转移才要特别注意,不过后者基本上不用,我们做了比较的,稳定性和重复性前者要好得多,当然对于高手来说也无所谓,但我觉得能做湿法的尽量还是用湿法。
1. 这种情况下我们通常会同时跑两块胶(或者一块胶切成两半),一块染色一块做Western,同步试验才能确保电泳没有问题,不知道你是不是这样做的。
2. 转膜我们一直用的25mM tris 192mM Glycine 15%甲醇(两次一换),各个实验室会用一套比较成熟的系统,如果你们的系统一直很好我觉得你的buffer你也不用怀疑它,只不过要注意check一下你的PH值。一旦摸索出较好的系统后不要轻易更换所使用的试剂厂家。
3. “考马斯亮蓝染色,干干净净,PVDF膜用丽春红染色,也什么也看不到”
的确,73KD这种条件下不会转过,所以我觉得是不是你的电极插线或者是做三明治的夹具放反了,全部转到缓冲液中去了,滤纸不会有什么问题,两种都可以,我们在没有厚的专用滤纸前一直用的是3张普通滤纸。
4. 没有用过的抗体最好有阳性对照,甚至我们每次做都会使用阳性对照,确保我们在出了问题能迅速找到原因
如果手头没有什么成熟的western体系可以向各个做抗体的大公司索要,这些体系都非常常熟,自己练习两次就熟悉了。
==========================================================================================================
我们实验室都是用半干转,重复性,稳定性都还很不错的说,只要不是人粗心的(电转液配错了,误把电泳液当成电转液用)结果,是不会轻易短路的,再说半干转省事,至少不用冷冻啊,呵呵,一般恒流80mA,1h-2h都可以,我试过,感觉一个半小时比2个小时还稍微好点(预染marker为证),还省时间,对了,最好用3mm厚的滤纸,怕转干了。再就是我们电转液是不重复使用的,因为每次用的也不多,用2个10cm直径的表面皿装转移液就可以了
作者:
tie8
时间:
2013-10-21 15:17
严重同意蛋白的重要性,actin的WB正常,并不代表sample合格。
我上个月做WB做了半个月,目的条带就是出不来。后来改做ACTIN,很容易条带就出来了。但换回目的蛋白的一抗还是出不来。开始怀疑是一抗的问题,换了一瓶一抗仍没有效果。后来同一个膜同时跑了 actin和目的蛋白,同样是actin条带出的很好,目的蛋白出不来。
后来考虑是sample的问题,重新提取了蛋白,用同样的方法做,目的蛋白很明显就出来。
所以,actin能出来,并不代表sample里的蛋白没有降解,尤其是目的蛋白含量不高的时候。
作者:
uuooii
时间:
2013-10-21 15:18
我就跑胶和转膜谈一点儿自己的体会
1、跑胶 电泳Buffer重复利用的时候要注意,不能混浊,有时候能用个3-4次,有时候第二次都不行了,事先前预电泳一下,看看气泡的均匀度;Buffer一定要淹过梳子孔,否则就会发现今天的蛋白不会跑;跑的时候先用低电压跑过浓缩胶,再换高电压跑分离胶,不要追求速度,慢慢跑条带会分离的更好,特别是对于高分子量的目的蛋白;
2、转膜 我用的是湿转,Buffer一定要预冷,最好提前一天配好放4度;滤纸不要大过PVDF膜,防止短路;胶在负极,膜靠近正极,放入Tank前检查一遍会使你免以体会放反的痛心疾首;膜要标记好正反面;转膜过程中,经常过去看看,防止意外,如电泳仪接触不好。
就这些了,请各位指正
=======================================================
transfer buffer可以在电泳上完样之后直接丢到-20度,等电泳完了就差不多了
滤纸我们天天都用大于PVDF膜的,不会短路。
作者:
DNA
时间:
2013-10-21 15:24
我最近一直在做,可是碰见一下问题向高手们请教,希望提点改正意见:
1 转膜时,多次出现小分子量的转膜效果不如中分子和大分子量的(用的彩虹MARKER,但14KD的亮黄色的条带仍然在胶上,转膜后的胶染色后小分子量区域的蛋白残留较多。在整张和将不同区域的分别转膜时均出现这种情况。)
2 在暗室里肉眼可以看见发光,可是胶片上一无所有。
3 ECL没有表达,可是用DAB时膜上却出现条带。
4 手工显影时,胶片的显影时间和温度有很大关系吗?显影的时间掌握如何比较恰当?
==========================================================================================================
dab可以显出目的条带说明流程没有问题。
而ecl敏感性应该更高反而没有出结果,表明问题出在化学发光法显色上。
A液和B液放置过久,里面所含过氧化氢可能会分解,请考虑是否问题出在这里,另外考虑显影液和定影液,以及显影时的操作时细节是否正确;DAB显影的话,应37度,ECL应该和温度没多大关系;DAB在37度显影一般5分钟内就可以出结果,ECL显影时间参照说明书。
作者:
jrwyyplt
时间:
2013-10-21 15:24
求教各位高手,最近我的Western blot经常是出来干干净净,什么都没有,成功率低。经过SDS-PAGE胶考马斯亮蓝染色,证明电泳没有问题,蛋白也没有降解。用正对照蛋白做点杂交也证明一抗,二抗以及发色系统没有问题。
疑点就集中在转膜过程上,因为之前试验正对照蛋白(30KD)的时候用的转膜用的厚吸水纸用完了(“三明治”两侧各一张),换成了薄的滤纸(两侧各三张),自从换了这个以后就一直有问题了,即使正对照也很难做出来。我用的是湿转法,转膜buffer同《分子克隆》上:48mM Tris,39mM 甘氨酸,20%甲醇,0.0375%SDS,转膜条件:4度,200mA稳流一小时,我的蛋白比较大,73KD,这个条件是否合适?转膜结束之后将胶用考马斯亮蓝染色,干干净净,说明已经转走了,将PVDF膜用丽春红染色,也什么也看不到,感觉不太可能转过头,因为之前30KD的蛋白用这个条件也可以的。最近我又将转膜buffer的甲醇比例下调到15%,其他条件相同。结果也是一样失望,各位有什么建议么?
==========================================================================================================
我也碰到相同的问题了.最近我的蛋白老是转膜不上.已经连续做了3次了,我的蛋白是72Kd的.使用的电流是不超过300mA,电压是110V,转膜时间是1.5h,结果3次都是发现预染Marker没有转上NC膜,同时胶上的预染Marker也没有了,再对胶进行考马斯兰染色,和对膜进行丽春红染色都没条带,但是在做预实验的时候做成功的,当时也出了小插曲就是发现没有转上进行胶的考马斯兰染色见到条带,随后再进行了一次转膜就上去了.
现在不知道是电流电压的问题.还是什么其他的问题,请教有经验的前辈们指教一二,
今天开始准备同时样品跑2块胶.一块就做考马斯兰染色.一块电转.准备用12v低温4度冰箱电转过夜.再试试.如果还没转上.而考马斯兰有条带,就准备再用染色的胶转一次.真的不知道究竟湿转的电流电压和时间是怎样的.请有作过的给个建议!!
急等!!谢谢!!
作者:
jrwyyplt
时间:
2013-10-21 15:25
今天我的转上去了.嘿嘿
我调整了一下方法,一个就是三明治的那个海绵变薄了.改用厚的和新的.然后就是使用的夹层滤纸用5层,保证夹子夹紧.还有就是在做三明治前一定要用力挤干净海绵中的空气然后再充分浸入到电转缓冲液中.保证没有空气,还有就是湿转的时候短路问题基本可以不要考虑.记得我原来转膜成功的电压就是110v电流差不多是270-300mA之间,1.5h,昨天我用的是12v恒压转膜过夜电流在35mA-40mA之间,为了放止转膜过头使用了2层NC膜,结果2层上都有预览Marker转上了.嘿嘿.同时做的一块胶进行的考马斯兰也见到有目的蛋白条带.
实验好痛苦啊,浪费的一周时间还是比较值得的.
仅供各位参考!
另外我的蛋白是72kD的估计今天晚上可以有得到ECl的显影图了!!
作者:
xue258
时间:
2013-10-21 15:25
我们WB做过很多次,一直以来条件都很稳定,可现在不知道是天气变冷了还是怎么了,在ECL显影时,最初两三分钟条带还能肉眼看到,但很快就消失了,X光片显影也没有,请问各位高手这是什么原因呢?我们根据说明书得解释,尝试改变二抗的浓度,已经不同浓度都试过了,还是老样子,请问各位高手,指点一下???谢谢啦
作者:
nut6694
时间:
2013-10-21 15:26
QUOTE:
原帖由
xue258
于 2013-10-21 15:25 发表
bbcodeurl('http://bbs.antpedia.com/images/common/back.gif', '
%s
')
我们WB做过很多次,一直以来条件都很稳定,可现在不知道是天气变冷了还是怎么了,在ECL显影时,最初两三分钟条带还能肉眼看到,但很快就消失了,X光片显影也没有,请问各位高手这是什么原因呢?我们根据说明书得解释,尝试改变二抗的 ...
一:确定问题出在显影上?
重心检查过程,只家2抗一样能和ECL反应。
二: 加2微升过氧化氢试试,ECL放置过久过氧化氢会分解。从而影响显影。
三: 你们曝光多久?
25min试试。
不能完全照搬说明书
作者:
xue258
时间:
2013-10-21 15:26
同意楼上的我原来肉眼能够看到那些亮的条带.现在没有了,而且原来显影2-5分钟很浓了.到30分钟反而不浓.淬灭了.现在2-5分钟基本没有条带.要压片1小时才可以看到条带.
可以延长压片时间!!
作者:
owanaka
时间:
2013-10-21 15:27
" 滤纸不要大过PVDF膜??哎,苍天啊,大地啊,到底滤纸,膜.胶的大小顺序是什么啊 怎么每个人说法都不一样啊 "
就我们实验室的情况来看,这三者之间没什么很大的讲究.这三者的大小关系并不会决定你的实验的成败.
作者:
vera+
时间:
2013-10-21 15:28
我做了很久的WB,最大的一个难点感觉在于样品制备上!蛋白的降解有些时候是你想象不到的,可能出奇的糟糕,也可能完全没事~~如果单独做WB的话,感觉以“快速彻底裂解,先变性后保存”的原则比较有效,尽量选择足够强的裂解液,甚至直接给loading buffer裂解,然后取出部分蛋白定量,其余加入loading buffer100度充分变性,再分装保存。有些时候比蛋白酶抑制剂更有效。
另一个感觉就是样品中目的蛋白的量,限于WB的检测下限问题,一般目的蛋白量最好别低于1ng(虽然ECL法下限是0.1ng,即100pg),最好在10ng以上为好。这就要求在做WB之前必须充分考虑目的蛋白的可能丰度,最好充分的准备工作然后再设计细胞量或者组织量,千万别把样品搞稀了,否则就不好办了。
最后,还是多看文献,在做某个蛋白之前最好看看别人是如何做的,有什么特殊要求,内参如何选等等。当然也包括多来丁香园学习~~:〉
=============================
战友讲的好啊!!!。。。。。。
蛋白提取第一重要!第二就是抗体了!
作者:
30moonriver
时间:
2013-10-21 15:28
请问我做的是蛋白差异表达,但是现在做出来的结果是感觉不同组织的差异不是很大,可能是目的蛋白都显得深了,请问能有什么方法使目的蛋白显出来但又不是很深?
我自己想,一抗二抗的浓度和时间有没有关系?还是一抗的关系不大,而二抗的关系大?曝光的时间应该有关系,还有就是是不是加样量大一点差异会明显一点?
请问有没有高手能做较为系统的阐述?谢谢!
作者:
wood533
时间:
2013-10-21 15:29
我的蛋白是多肽,只有40个氨基酸,分子量也就大概几个kd,
请教各位,象这种蛋白在做western时该注意什么?转膜用半干还是湿转好呢?
作者:
summerxx
时间:
2013-10-21 15:30
请问我做的是蛋白差异表达,但是现在做出来的结果是感觉不同组织的差异不是很大,可能是目的蛋白都显得深了,请问能有什么方法使目的蛋白显出来但又不是很深?
我自己想,一抗二抗的浓度和时间有没有关系?还是一抗的关系不大,而二抗的关系大?曝光的时间应该有关系,还有就是是不是加样量大一点差异会明显一点?
请问有没有高手能做较为系统的阐述?谢谢!
===========================================================================================================
1。蛋白上样量是多少,一般情况下,20-30ug总蛋白已足够了。
2。曝光/显影过深会掩盖掉蛋白浓度的差异,提高一抗或二抗的稀释度可以解决这个问题。
3。至于曝光时间,用Amersham ECL底物检测,如果曝光时间少于几十秒就可以显示出明显条带的话,说明蛋白量或一二抗过饱和了,可以减少用量。
作者:
summerxx
时间:
2013-10-21 15:30
我的蛋白是多肽,只有40个氨基酸,分子量也就大概几个kd,
请教各位,象这种蛋白在做western时该注意什么?转膜用半干还是湿转好呢?
=====================================
1。高浓度PAGE胶(15-20%)。
2。0.22um转印膜。(一般使用的是0.45um)
3。转移buffer中甲醇含量不要太低(15-20%)。
4。凝胶在转移buffer中平衡时间不要超过10min。
作者:
summerxx
时间:
2013-10-21 15:31
我们WB做过很多次,一直以来条件都很稳定,可现在不知道是天气变冷了还是怎么了,在ECL显影时,最初两三分钟条带还能肉眼看到,但很快就消失了,X光片显影也没有,请问各位高手这是什么原因呢?我们根据说明书得解释,尝试改变二抗的浓度,已经不同浓度都试过了,还是老样子,请问各位高手,指点一下???谢谢啦
=============================================================================================
会不会是显影液温度太低?洗照片时显影液温度是挺关键的因素,洗胶片可能也是如此,我们实验室有空调,没遇到过这种情况。
条带肉眼可见,反应太强烈了,容易淬灭。
作者:
fox_79
时间:
2013-10-21 15:31
成败的经验俺也有不少,样品制备、电泳转膜上抗发光前面各位都说了很多,俺就拿显影后的条带异常来说:
1 背景深,可以看到泳道 多数的可能是上样品太大或者抗体用量过多导致,按照一般的推荐,总蛋白量控制在50-100ug左右,抗体稀释度(尤其是一抗)需要做一个优化
2 条带拖尾,最大的可能是样品溶解不佳,要根据样品选择合适配方的裂解液保证样品充分裂解,上样前离心去除不溶物,曾经在样品上面载过大跟头
3 条带模糊,这个最复杂,可能原因是多方面的,包括胶本身的原因,缓冲液的原因SDS质量原因等,但我遇到过的是还原剂β-ME的原因:由于loading buffer存放时间过长β-ME挥发浓度降低,不能把二硫键完全打开,导致条带模糊,后来把β-ME现用现加,条带就好了
4 传说中的微笑现象,就是边缘的条带位置高中间低的现象,我在夏天温度高时遇到过,查过一些资料,说是因为散热不均造成的迁移率不一致,电泳的时候电压不要过高或者直接把电泳槽放在在盛有冰水的盆里就能解决
作者:
windy+++
时间:
2013-10-21 16:03
我觉得做western 应该注意以下问题
1、 首先保证目的蛋白的表达,蛋白样品处理、跑胶,每个实验室都有一套经验,应该没有问题
2、关键的是转膜,电流、时间要根据自己实验室的转膜设备和以前做过的经验自己找合适的条件
3、再就是一抗、二抗的问题了,一定要保证肯定他的来源和质量
作者:
zhenxin
时间:
2013-10-21 16:13
大家好,我现在做的目的蛋白是280kd的,组织,用的ripa裂解液,裂解的时候只加了PMSF,结果染了PAGE胶后连阳性对照也没有条带,是因为蛋白酶抑制剂不够吗?
作者:
flower-201
时间:
2013-10-21 16:14
根据你所说的应该考虑蛋白降解,首先注意取组织的过程、组织提蛋白的过程,还可以加COCKTAIL试试。
作者:
zhenxin
时间:
2013-10-21 16:23
谢谢楼上的!!
我从刚托臼处死的大鼠取出脑组织后,放入-80,10分钟后取出在匀浆,可以吗?
另外,一般匀浆的时间多长?如何看出匀浆好了?
作者:
nut6694
时间:
2013-10-21 16:24
大家可以多讨论讨论磷酸化蛋白的WB方法
作者:
kuohao17
时间:
2013-10-21 16:24
偶有个问题,至今一直不明白。
用天根的marker,在小梳子制孔时跑的还可以,
但在大梳子时就只能跑出一半的宽度,就是只有加样孔的一半宽,而且跑很难看,这是为什么?
用其他marker从未出现过这种情况。
作者:
ukonptp
时间:
2013-10-21 16:28
偶有个问题,至今一直不明白。
用天根的marker,在小梳子制孔时跑的还可以,
但在大梳子时就只能跑出一半的宽度,就是只有加样孔的一半宽,而且跑很难看,这是为什么?
用其他marker从未出现过这种情况。
======================================================================================================
我也遇到过的,由于两侧lanes的样品加样可能偏多,会把marker向中间挤压的,形成变窄或不规则的泳道.可以稍微减少上样量试试.呵呵.
作者:
2541
时间:
2013-10-21 16:29
这种可能是存在,我具体情况没说清楚。
我的上样量只有30ug,对大孔来说绝对不多,而且我同时用了另外一个marker,条带就很漂亮。天根的就只有一半加样孔的宽度。
不知道怎么回事。
作者:
eric930
时间:
2013-10-21 16:29
如果同样条件下天根的跑不过其它marker,那只能是marker本身的问题了.请高手指点下呢,呵呵。
作者:
PCR
时间:
2013-10-21 16:29
请指教一下,
我做的心肌组织蛋白,共做了两张膜,除了一抗不一样外,其余条件完全一样(一个是actin,一个是troponin),结果很奇怪,actin那张膜上,背景清楚,但是条带很浅,有些孔看不见(我作了一个浓度梯度,6,3,1.5mg/ml).但是troponin那张膜上,背景比较重,非特异条带多得我都着不到目的条带了.我大致得条件是浓缩胶60V,分离胶100V,转膜350mA55分钟.ECL
我的一抗是SANTA CRUZ(两个都是1:1000)是不是不好?我的膜杂带多是不是也说明我的蛋白是有的,就是提蛋白,电泳,转移都没有大问题,但是为什么actin为什么会这么少?
作者:
PCR
时间:
2013-10-21 16:30
还有,抗体一般的使用期限是多久?
作者:
nn255
时间:
2013-10-21 16:30
我也作了好长时间western , 一直有一个疑问,为什么我的western老师不稳定呢
ECL曝光老是一点亮光都没有,但有时候同样的条件,发光又很好
究竟是什么原因阿 百思不得其解
作者:
jkobn
时间:
2013-10-21 16:31
我也作了好长时间western , 一直有一个疑问,为什么我的western老师不稳定呢
ECL曝光老是一点亮光都没有,但有时候同样的条件,发光又很好
究竟是什么原因阿 百思不得其解
==========================================================================================================
实验重复性有很多影响因素,每个环节每个细节都可能影响结果,在规范操作保证人为因素尽量一样的情况下,还应该注意的是一些试剂可能随着环境的变化效率或作用结果可能有改变,应该严格注意温度,PH等。
祝好运!
作者:
sr9971
时间:
2013-10-21 16:31
还有,抗体一般的使用期限是多久?
==========================================================================================================
我们实验室的经验是稀释一抗使用PBST+NaN3+BSA,进口原装抗体一般10次左右(或者更多)都没什么问题,但国产或者分装抗体要看运气如何。
作者:
c86v
时间:
2013-10-21 16:32
成功过的战友讨论下,你曾经WESTERN做不出来是那部分出问题了.看看各部分出问题的几率,为还没有成功,仍然苦苦寻找结果的战友提供点参考(由于本人经验有限,不足之处还请斑竹和各位大侠改正):
1、蛋白提取问题,蛋白降解,或者上样量少导致检测失败。
2、蛋白电泳失败。
3、转膜问题:转膜不充分,过转,温度过高导致蛋白结构改变等。
4、抗体问题。
5、显影问题。
6、其它。
请战友提供各部分问题的主要说明和对策,谢谢!祝丁香园蛋白版越来越旺!
=====================================================================
我跑完电泳用考马斯亮蓝染胶再用甲醇冰醋酸脱色,胶上什么都没有,什么原因呢?请大侠赐教!
作者:
nn255
时间:
2013-10-21 16:33
可以用考马斯亮蓝染胶 6h以上,然后水煮三遍看看,如果还是没有条带,就是蛋白样品的制备有问题.
作者:
c86v
时间:
2013-10-21 16:49
请问楼上的:
"水煮三遍":是在染色液里煮胶吗?每次多长时间,间隔呢?
谢谢!!!
作者:
JK.jon
时间:
2013-10-21 16:50
大家可以多讨论讨论磷酸化蛋白的WB方法
==========================================================================================================
我贴一个。
做过CREB-p水平检测,与测CREB最大不同之处在于保持CREB的磷酸化状态。我用的细胞裂解液中都含有氟化钠(NaF)和焦磷酸钠(NaPPi2) 两种成分 ,用来稳定CREB-p。因为检测creb-p的单克隆抗体很可能就是识别CREB与p结合部位的结构的,如果CREB-P在处理过程中降解,最后必然检不出来。
我看过的几个报阴性结果的文献中,其样品处理过程多数没有考虑保护磷酸基,这可能是失败的原因之一吧。
附上相关文献一篇:ZHao LX,Brinton RD.J Neurosci,2003,23(10):4228-4239.
作者:
qumm1985
时间:
2013-10-21 16:51
我也一直在做WB,体会是一抗最关键!
其实有些蛋白特别是重组蛋白不用纯化一样可以直接上样,用量不宜太多,对照电泳带能看见就行,这样还能避免本底高的问题.如果一抗用单抗,则封闭不宜过长,一抗如果效价不高,可以适当延长反应时间,例如4度过夜.至于二抗和显影都比较常规,只是注意每步要好好洗膜,把非特异降到最低.另外膜也注意步要损伤,否则会影响显影.
作者:
windy+++
时间:
2013-10-21 16:51
我的蛋白上有biotin,所以我只需要用到可以检测用的avidin就可以了,不用一抗,二抗。问几个很菜的问题:
1. 我想知道使用streptavidin-horseradish peroxidase做chemiluminescence检测的设备是什么?我们实验室只有一个扫描胶用的普通扫描仪,是不是不能用来做chemiluminescence检测?如果不行,有没有什么便宜的解决办法?
2. 请大虾们给推荐一个protocol
作者:
junjie05
时间:
2013-10-21 16:52
我是个做western的新手,已经做了几次,均无结果。我觉得这些过程中最关键的就是蛋白的提取,一定要先有东西然后才能检测它。象我做膜蛋白,用的是提取的细胞总蛋白,做了几次western都没结果,现在也不知道到底我的蛋白有没有提取出来,还是别的方面有问题。真的很郁闷。所以,我觉得必须要保证你的蛋白提取是有效的,否则后面做的一切都是白费。
作者:
nut6694
时间:
2013-10-21 16:52
QUOTE:
原帖由
junjie05
于 2013-10-21 16:52 发表
bbcodeurl('http://bbs.antpedia.com/images/common/back.gif', '
%s
')
我是个做western的新手,已经做了几次,均无结果。我觉得这些过程中最关键的就是蛋白的提取,一定要先有东西然后才能检测它。象我做膜蛋白,用的是提取的细胞总蛋白,做了几次western都没结果,现在也不知道到底我的蛋白有没有 ...
蛋白制备确实非常关键,建议你在样品处理过程中可以试着不煮沸变性,加了LOADING BUFFER直接做一下看看。或者专门有提膜蛋白的KIT,针对膜蛋白水溶性不强的特点可以比较有效得提取较高丰度的样品。
作者:
INK
时间:
2013-10-21 16:53
我来说一下吧,平时做的挺多的,还是有点心得的。。。
第一,提取蛋白的问题:我不知道你们用的是甚么方法来提取蛋白的,基本上提取组织蛋白和细胞蛋白的方法都很成熟,所以我觉得这不是主要问题,关键无非就是浓度低点罢了,但是western的分辨率很高,不会影响到作不出来的。为了防治这方面的问题,可以设置阳性control。
第二,跑胶的问题的可能性就更小了,反正从我平时做的情况来看,这个几率是零,因为整个过程都在肉眼监视下操作,即使发生问题也很容易及时发现。倒是值得一提的是下胶的时候一定要小心,不然把胶弄坏了才叫心疼,整个重新来过,哭。。。。。
第三,转膜是个关键!!!首先转膜时间和电流需要根据你目的蛋白大小以及转膜方法,还有膜的材料等等因素相关联,具体需要慢慢摸索,也可以向你实验室做的好的师兄师姐请教,我就不赘述了。需要提醒一句的是:做小蛋白的时候一定要注意!!!因为它很容易降解,所以下胶和转膜之间的时间越短越好!切记!!
第四,抗体问题当然也是很关键,但是无论是商品化抗体还是自己打得抗体也好,再做之前都会经过检测的,所以出现问题的可能性也不大,除非你马虎大意地加错了,如加错二抗(我就犯过这个问题,把鼠的加成了兔的)!切忌!也可以通过对照来排除。。。
第五,显影出现的问题也不太常见,可以问问用过这批显影液的同实验室的人来确认。
其余可能出现的问题也很多,但是都是低概率事件,归根结底都是粗心大意造成的,因此状态不好的时候建议不要做试验,尤其是western这种时程很长的试验,一步有错,前功尽弃。。。。
稍微总结一下,我的观点就是转膜那一步最重要,一定要小心。。。。
希望有所帮助,呵呵
作者:
tangxin_80
时间:
2013-10-21 16:53
内参做出来,而目的蛋白大部分做出而有个别的做不出来不妨考虑一下几个因素:
1 仔细再阅读一下抗体说明书,单抗还是多抗,如果单抗是来源小鼠的还是兔的(CST公司有很多兔的单抗),如果是兔的单抗,二抗选择时就要用抗兔的多抗。
2 看看分子目标分子量的大小,确定胶的浓度,本人在做凋亡方面深有感触就是做Caspase的小片段,用15%、12%的胶就能做出,而10%的就做不出来。
作者:
kulee
时间:
2013-10-21 16:54
请教高手:
分子量120kd的表达蛋白,8%分离胶,5%浓缩胶,电泳100v,2.5h;转膜恒流125mA,2h,此条件是否可以;另外,表达量较低,有什么好方法提高灵敏度呢,先谢谢啦
作者:
nut6694
时间:
2013-10-21 16:54
QUOTE:
原帖由
kulee
于 2013-10-21 16:54 发表
bbcodeurl('http://bbs.antpedia.com/images/common/back.gif', '
%s
')
请教高手:
分子量120kd的表达蛋白,8%分离胶,5%浓缩胶,电泳100v,2.5h;转膜恒流125mA,2h,此条件是否可以;另外,表达量较低,有什么好方法提高灵敏度呢,先谢谢啦 ...
蛋白电泳时浓缩胶和分离胶的电压不一样,浓缩胶可以用80-100V,跑进分离胶后用160-200V跑.
转膜你是用湿转还是半干转?根据我的经验,半干转条件,恒流1-2mA/cm2,转2-2.5小时.如果湿转,用恒压100V转2.5小时.你试试,再根据转膜效果调整一下.
如果表达量低,可以通过浓缩样品蛋白,加大上样量等调节,也可以用比较灵敏的显影底物.
作者:
xingyi08
时间:
2013-10-21 16:55
我们电泳时一般是浓缩胶90v,分离胶120v,或者浓缩胶60v,分离胶90v;当然,200v跑全程也可以。不过建议用低电压跑,这样发热低,条带会好看一些。大分子量蛋白转移时,试试加0.01%-0.05%SDS,看有没有好转。
蛋白表达量低,可用尽可能少的lysis buffer收蛋白,以提高浓度,加大蛋白上样量。尽量避免用浓缩管等手段浓缩蛋白样品,很难保证活性蛋白成分在这一步处理中不发生变化。
提高灵敏度的一个方法是换ECL显影试剂,Amersham的ECL、ECL plus、ECL advance的检测灵敏度有几何级数的差别,可以试试。但用plus或advance时,一、二抗的稀释度要比ECL时高得多,否则,背景会高得震撼住你的:)
===========================================================================================================
请教高手:
分子量120kd的表达蛋白,8%分离胶,5%浓缩胶,电泳100v,2.5h;转膜恒流125mA,2h,此条件是否可以;另外,表达量较低,有什么好方法提高灵敏度呢,先谢谢啦
作者:
JK.jon
时间:
2013-10-21 16:56
还有PBS放置时间过长,长菌导致抗体失效,DAB失效也是原因之一。
作者:
shenkunjie
时间:
2013-10-21 16:58
试验没有进展,老板着急,我心里更急。望高人指点。
我做的是心肌组织蛋白,我已经作出了目的条带,以及actin,但是我发现actin出来后粗细不等,说明我测蛋白的时候有问题,于是重做。我用的bio-rad的DC蛋白测定试剂盒,我以前做的浓度大多有8-10mg/ml,我准备将放在-80的蛋白拿出来融解后再测,发现样品溶解后有一些沉淀,于是我将其在4度离心后再取上清再测,发现蛋白浓度降了很多,只有2-3mg/ml。那些沉淀出来的蛋白到底是甚么,会不会影响我最后的结果?蛋白浓度降了这么多,会不会对我的目的蛋白有一些损失?那里标本可是来之不易啊,怎么办?
希望很多人能看到这个帖子,给点建议!
作者:
glass
时间:
2013-10-21 16:58
QUOTE:
原帖由
shenkunjie
于 2013-10-21 16:58 发表
bbcodeurl('http://bbs.antpedia.com/images/common/back.gif', '
%s
')
试验没有进展,老板着急,我心里更急。望高人指点。
我做的是心肌组织蛋白,我已经作出了目的条带,以及actin,但是我发现actin出来后粗细不等,说明我测蛋白的时候有问题,于是重做。我用的bio-rad的DC蛋白测定试剂盒,我以前做的 ...
考虑如下:
1. 是否由于测定时或上样换算体积时疏漏
2. 放在-80度的蛋白时间有多久? 大于2个月最好重新提
3. 样品溶解后出现沉淀,可能是提取蛋白时候没有充分裂解;另外一种可能是细胞骨架蛋白的聚集物.可以考虑0度12000g离心5min,此时取上清定量
4. WB的检测下限是1-5ng,所以应该没有问题,可以考虑染胶染膜看看效果
5. 一般离心应该不会影响蛋白浓度,毕竟体积没有发生改变
作者:
shenkunjie
时间:
2013-10-21 16:59
我离心后下面是有沉淀得,所以细胞浓度会下降。如果是细胞骨架蛋白得话,那我得actin就可能会不一样粗哦?0度,12000,5min可以吗?我想下次做的时候14000,4度,10min?不知哪个更好?
作者:
cocacola
时间:
2013-10-21 16:59
transfer buffer可以在电泳上完样之后直接丢到-20度,等电泳完了就差不多了
滤纸我们天天都用大于PVDF膜的,不会短路。
========================================================================================
一般所用的pvdf膜都不会大过胶的,只要滤纸没有比胶还大,应该都还ok拉,不大会出现短路这种倒霉的事情吧!:)
作者:
greenbee
时间:
2013-10-21 17:00
我的蛋白样品浓度为1.2mg/ml,样品上样量为20,样品是制备好直接上样,可是显色后目标条带非常浅,后来用新的一抗、二抗结果一样。可是以前几次蛋白浓度只有0.2mg/ml时,目标条带都非常清晰,请各位高手指教。
=====================================
我觉得你的蛋白可能是降解了。不然不会差这么多
作者:
greenbee
时间:
2013-10-21 17:00
个人感觉做wb首要的一步也是最关键的一步就是提蛋白,相对于细胞蛋白来说,组织的蛋白更难提取一些(蛋白的纯度和降解方面不易控制),上面的战友也提到了组织匀浆器和玻璃匀浆器迅速冰上进行的方法,个人感觉为避免蛋白的降解最好采用液氮研磨的方法,组织块不用剪的太碎也可以,研磨过程中注意补充液氮就可以了,不受时间的限制,唯一的缺点就是对组织有一定的损失。
===========================================================================================================
同意以上观点,我们在做组织匀浆的时候,是先在液氮下研磨,然后加入裂解液,将研钵放在冰上,稍微融化一下,然后用杵将研钵中的裂解液与组织充分混匀。然后再将组织液转移到匀浆器中,在冰浴中继续匀浆。我觉得这样的效果比较好。希望多多交流。
作者:
qumm1985
时间:
2013-10-21 17:00
过一段也要做western了,上来向大家学习.
不过有的地方看不懂,
大家说多看书好
还是直接去向实验室的师兄试姐们请教的好?
作者:
H2O
时间:
2013-10-21 17:00
我觉得如果蛋白含量丰富,一般很容易做出来,但如果样品含量极低,要方方面面都考虑齐全了,技术成熟了才能做出来.对于新手,建议先易后难较好!
作者:
阿k
时间:
2013-10-21 17:01
其实大的步骤大家应该都没有问题,我说说细节的问题!
1、蛋白的提取:组织的蛋白由于比较杂,不太好做,细胞的相对好做些!蛋白提取时一定要注意在冰上操作,组织的提取我直接研磨后取上清(什么也没有加,也可以出来!)。buff处理煮沸后冻存,使用的时候一定要再次煮沸,因为蛋白有可能在保存的时候复性!
2、配胶:不论基层还是分离胶一定要注意给足充分的聚合时间(一个小时),否则胶聚合不均匀跑出来的是扭曲的条带!TEMED一般不会出问题,AP一定要新鲜的一周之内的(使用时闻闻,如果有刺鼻的气味就说明没有过期)!
3、封闭:封闭的时间最好常一点,我们采用室温摇床4h,出来的背景非常干净(超敏发光液的也很干净)!
4、一抗:个人感觉非常重要,抗体经量买国外的(并没有贬低国产的意思),使用时一定要分装,多次冻融后抗体很容易降解!孵抗体的时间最好4度过夜,冰箱取出来后室温在放置1h。(我们实验室几乎不用封闭袋是用的湿盒,是把膜平铺在蜡板上表面滴上抗体,要表面张力的那种,非常节省抗体!)
5、转膜也很重要:我们采用湿转,湿转不容易把蛋白转过!转膜时一定注意温度,在转之前把电转液冰箱里冻着,使用的时候在加!另外转移槽外面要加冰块冷却,很多情况下由于问题过高会导致效果很差!
6、洗膜:不要怕浪费TBST,也不要为了省时间少洗!我们都是15min,4次!出来的背景很好!
7、发光:发光液配好后用小盒子装,把膜泡在里面30s,要全部浸在里面,直接上凝胶成像仪。(发光液直接滴在膜上的经常会发光不均匀)!
以上是一点心的,欢迎大家讨论!
作者:
shenkunjie
时间:
2013-10-21 17:01
我做了不少次WB,失败的时候占多数,汗,有很多失误的地方,最主要的影响因素还是一抗问题,一抗浓度要合适,我最近做跑GRP78,总是不出来,很淡,做了几次才明白一抗有点失效了,郁闷之极。还有一些细节问题,比如剪膜,显影时间等...相信大家都遇到过。
作者:
BOSS2011
时间:
2013-10-21 17:02
呵呵我也来说两句!刚做WB的时候,丁香园的一个前辈曾说:WB最关键的步骤是(1)蛋白提取;(2)电泳;(3)转膜;(4)显影。后来实验过程中发现这几个点真的很重要!好了,我来说说我的感受:
1、蛋白提取问题。
一般的蛋白提取,按照标准的操作应该问题不大,组织蛋白的降解没有我想像的那么“脆弱”。我们在-80度保存将近1年的组织,也可以做出来结果,当然还是尽快提取蛋白最为保险!但是,针对核蛋白、膜蛋白或者线粒体蛋白等特殊的蛋白,采用专门的裂解液是很有必要的,蛋白提取不好后面的步骤无法进行。蛋白提取后变性过程中,注意上样缓冲液的问题!以前我们购买的劣质缓冲液严重影响蛋白变性,导致考马斯亮蓝R-250染胶的结果很差,后来自己配置上样缓冲液就解决问题了。证明蛋白提取没问题(排除了其他的因素),是劣质缓冲液不能满足实验要求。所以这个问题也需要引起大家的注意。
2、蛋白电泳失败。
电泳我遇到的问题不多,主要是电泳液的ph值要保证,重复使用的次数也就2-3次,不要怕麻烦而耽误了后面的实验。
3、转膜问题:转膜不充分,过转,温度过高导致蛋白结构改变等。
转膜是最关键的,转膜的条件因为大家的仪器不同(即使是同一厂家同一款产品条件也会产生差异),很难有个同一 的标准,因此需要大家摸索。一般每个实验室都会有做过WB的前辈,只好参照他的条件自己再摸索。
转膜完毕立春红染色不是很可靠,没有染上的也可以做出来,建议大家坚定信心还是坚持做下去。
4、抗体问题。
主要是一抗!这个问题不好说!需要点运气,我们实验室有人使用420元的国产抗体做的效果很好,但我们不敢保证下一次他还会有这么好的运气。经济能力许可建议买好的抗体(就是价格高的抗体),可以提高效率节省时间,而且好的抗体其稀释比例不需要摸索很久,一般按照说明书的操作就可以了。注意二抗的稀释比例不能太高,否则背景会深,我们一般1:5000!我们一般固定二抗的稀释比例(1:5000),搭配不同稀释比例的一抗来摸索抗体搭配条件。
如果一抗是羊抗,背景可能会很高,我们采用10%牛奶封闭过夜后,背景比较干净,这个方法可以试试!
5、显影问题。
环境温度是个问题!冬天我们做内参,条带显影都不够黑,而且显影时间会延长!有意思的是,吹口热气可以让荧光亮一些!这个和温度有关!可以开空调提高显影室的温度。我们实验室有人把显影液实验水浴箱37度孵育一下,也可以帮助显影。
显影的时候,如果荧光肉眼可见,压片时间以秒计算。如果荧光似有似无,压片时间以分钟计算。这个还得实战体会摸索一下。
6、其它。
建议大家蛋白提取后先做考马斯亮蓝R250染色,初步判定蛋白质量。
建议大家无论何时都要先做内参,保证WB体系没问题,然后再调整转膜条件、抗体比例、显影时间的细节。
这两点都是磨刀不误砍柴的细节,希望大家先做!
作者:
INK
时间:
2013-10-21 17:02
严重同意蛋白的重要性,actin的WB正常,并不代表sample合格。
我上个月做WB做了半个月,目的条带就是出不来。后来改做ACTIN,很容易条带就出来了。但换回目的蛋白的一抗还是出不来。开始怀疑是一抗的问题,换了一瓶一抗仍没有效果。后来同一个膜同时跑了 actin和目的蛋白,同样是actin条带出的很好,目的蛋白出不来。
后来考虑是sample的问题,重新提取了蛋白,用同样的方法做,目的蛋白很明显就出来。
所以,actin能出来,并不代表sample里的蛋白没有降解,尤其是目的蛋白含量不高的时候。
===========================================================================================================
我做的是磷酸化的STAT3,活化后应该都往细胞核里跑了,但我提的是细胞总蛋白,做了之后目的条带没出来,actin是有的,是不是一定要提核蛋白才行呀,还是p-stat3 本身丰度就太低了呀,求救ing!~
作者:
bs4665
时间:
2013-10-21 17:02
我一开始做western是显影液有问题。片子发白,就好像没曝光一样。
作者:
queen
时间:
2013-10-21 17:03
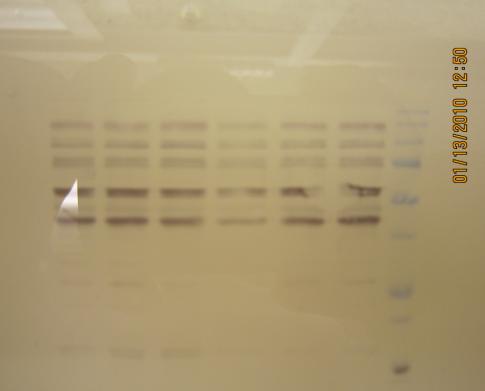
我感觉膜的处理和转膜的电压选择很重要。
我第一次做时,我的目的蛋白53kDa,实验室的technician给我的protocol中转膜条件是23v 30min。结果转完后一看胶上的marker条带还很清楚,后来我查了资料,改为80v 60min,效果很好。
我用的膜是PVDF膜,第2次做时没用methanol处理,结果膜上有一片透明的区域,当时不知为什么,后来在园里咨询了后才知道是没处理的原因,后来每次都用methanol浸泡2-5min,然后浸在转移夜里直到用时,效果很好。
贴张图大家看看区别。
图片附件:
30757292.snap.jpg
(2013-10-21 17:03, 11.5 KB) / 该附件被下载次数 5
http://bbs.antpedia.com/attachment.php?aid=19512
作者:
queen
时间:
2013-10-21 17:04
我感觉膜的处理和转膜的电压选择很重要。
我第一次做时,我的目的蛋白53kDa,实验室的technician给我的protocol中转膜条件是23v 30min。结果转完后一看胶上的marker条带还很清楚,后来我查了资料,改为80v 60min,效果很好。
我用的膜是PVDF膜,第2次做时没用methanol处理,结果膜上有一片透明的区域,当时不知为什么,后来在园里咨询了后才知道是没处理的原因,后来每次都用methanol浸泡2-5min,然后浸在转移夜里直到用时,效果很好。
贴张图大家看看区别。
图片附件:
93013458.snap.jpg
(2013-10-21 17:04, 8.75 KB) / 该附件被下载次数 1
http://bbs.antpedia.com/attachment.php?aid=19513
作者:
gogo
时间:
2013-10-21 17:04
刚刚开始做WB,第一次做的时候只是封闭的时间特别长,几乎有一天了(5%WB专用脱脂奶粉封闭)。第一次做的时候还出现了显色的条带(DAB+HRP显色),但是大小不对且对照和材料都有。后面做的时候就没有出现过条带了,但是PVDF膜上都会倍染成蓝色到蓝紫色的,不知道是怎么回事。
作者:
mysmdbl
时间:
2013-10-21 17:05
我用的是博奥森的抗体,多克隆的,目的条带44KD,封闭一小时,抗体4度过夜,二抗一个小时,可是除了目的条带以外,都出的了,目 的条带却一次都不出,请教各位高手,原因有哪些,如何解决?
作者:
www.1
时间:
2013-10-21 17:05
这个帖子怎么没人继续跟了,我做了将近一年western,最近遇到很诡异的问题,排除不了原因,但是也还是有点心得,跟大家分享一下
关于显影定影,我看前面的各位说的很少,实际上这个也很关键,我们实验室有很长一段时间做不出来,经过这种查找排除最后发现问题就出在这里
冬天天冷,ECL发光液跟二抗反应发光需要一定的温度,开始我们都是暗示直接压片,暗示没空调,大概3-4度的样子,一直做不出来,后来就把暗盒放到30度的培养箱中放一个小时
然显影液和定影液也预热了一下,出来的片就很好
后来我发现显影液和定影液温度太高和太低都会导致片子不清亮,还是要一定的温度,秋天的气温很适合
希望可以帮到大家
欢迎光临 分析测试百科 (http://bbs.antpedia.com/)
Powered by Discuz! 5.5.0
 图片附件: 30757292.snap.jpg (2013-10-21 17:03, 11.5 KB) / 该附件被下载次数 5
图片附件: 30757292.snap.jpg (2013-10-21 17:03, 11.5 KB) / 该附件被下载次数 5