 图片附件: 81960241.snap.jpg (2013-11-16 16:59, 49.33 KB) / 该附件被下载次数 6
图片附件: 81960241.snap.jpg (2013-11-16 16:59, 49.33 KB) / 该附件被下载次数 6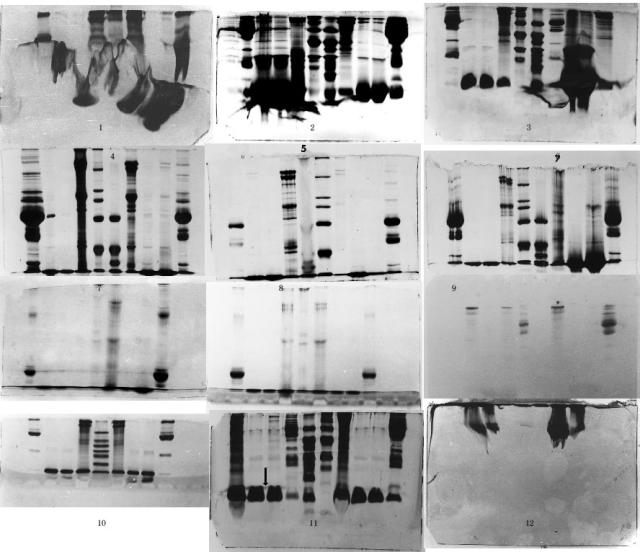 图片附件: 22110923.jpg (2013-11-16 16:59, 21.31 KB) / 该附件被下载次数 10
图片附件: 22110923.jpg (2013-11-16 16:59, 21.31 KB) / 该附件被下载次数 10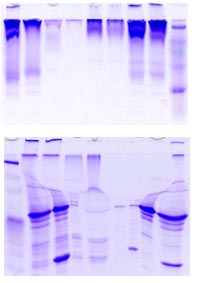 图片附件: 20252765.jpg (2013-11-16 17:24, 15.66 KB) / 该附件被下载次数 12
图片附件: 20252765.jpg (2013-11-16 17:24, 15.66 KB) / 该附件被下载次数 12 图片附件: 52375571.snap.jpg (2013-11-16 17:25, 22.88 KB) / 该附件被下载次数 10
图片附件: 52375571.snap.jpg (2013-11-16 17:25, 22.88 KB) / 该附件被下载次数 10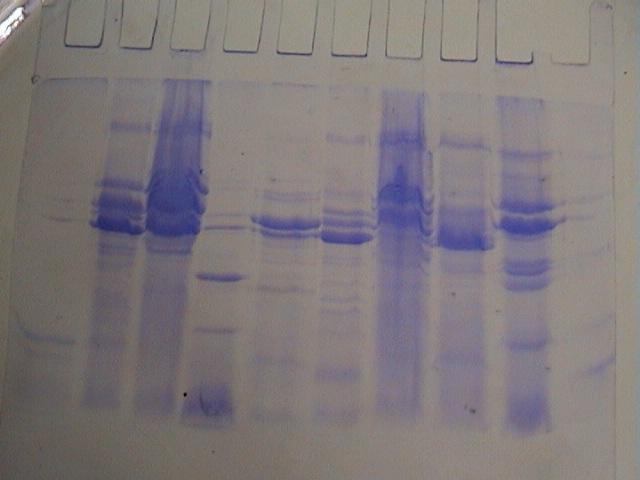 图片附件: 41520676.jpg (2013-11-16 17:32, 17.35 KB) / 该附件被下载次数 14
图片附件: 41520676.jpg (2013-11-16 17:32, 17.35 KB) / 该附件被下载次数 14 图片附件: 69153087.snap.jpg (2013-11-16 17:46, 49.14 KB) / 该附件被下载次数 11
图片附件: 69153087.snap.jpg (2013-11-16 17:46, 49.14 KB) / 该附件被下载次数 11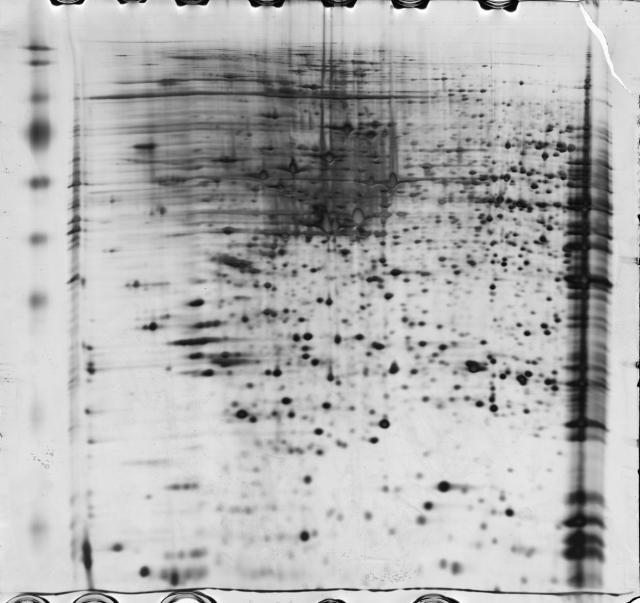 图片附件: 21835826.jpg (2013-11-16 22:10, 29.33 KB) / 该附件被下载次数 19
图片附件: 21835826.jpg (2013-11-16 22:10, 29.33 KB) / 该附件被下载次数 19
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |