这一点,不敢苟同,想与版主交流下。
这里面涉及到一个critical quality attributes的概念。多数抗体药物是九十年代末上市的,究竟哪些质控点影响临床疗效,哪些会造成免疫反应。在业内并没有公论。FDA只说,就事论事,CASE by ...
我还是认为,对于一个深入研究生物制药的人而言,这不是什么问题。这是一个基本问题。
因为我研究生物制药很少,也就根据自己看法,大概说一说。
如下指南和指导原则,如果大部分看了,应该对建立抗体类药物的思路,较清晰了。
1-cde《人用单克隆抗体质量控制技术指导原则2003》
2-EP7《monoclonal antibody for human use》
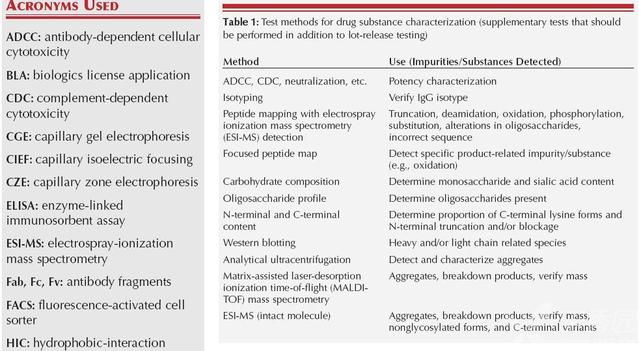
3-ICH Q6B《测试方法和接受标准:生物技术和生物药品》
4-FDA《Assay Development for Immunogenicity Testing of Therapeutic Proteins》
5-FDA《Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use》
怎么说呢,我认为如果把ICH Q6研究透彻了作者: qianqin1977 时间: 2014-8-6 22:22
简单说几句:
Protein concentration:通常采用分光光度法,前提是知道280nm转换因子。lowry法准确性和方法稳定性稍差。
Potency : 一般不能基于ELISA结果,至少得基于细胞学试验结果,流式细胞结合活性一般也不能完全代表Potency。
ID (Immunoassay):一般没有必要了,因为其他很多试验都间接代表了鉴别。
Sub-visible particles: 不溶性微粒,2010年版中国药典三部标准是不大于6000粒/瓶(不大于10um)和不大于600粒/瓶(不大于25um)
此外,如果作为药品,每年至少应该测一次N末端氨基酸序列。作者: 耗子=== 时间: 2014-8-6 22:26
这个只是产品的检测指标,检测方法buddesus 和horizon801的解释结合起来真是非常精彩,真没想到这个帖子揪出来这么多牛人。前段时间有人请教release lot test 是什么意思,呵呵,这个是不是很好的解释?
这些检测指标后面还有一大堆的东西,如何能够获得合格的产品还需要前期很多控制工作,我想这个应该是更加重要的,除了产品本身,细胞基质的检测以及控制也很重要。希望我们以此为基础,慢慢的摊开来,分别讲讲,我觉得比一个人闷头看文献好多了。作者: 阿k 时间: 2014-8-6 22:29
Sub-visible Particulate ---------------------- 29 particles/container ≥ 10 μm [no more than 6000]
这个不知道业内怎么称呼?求解。
次级可见异物?该纸包应该与溶解性和澄清度相关。
另外,楼主公布的只是你公司所作抗体项目的检测项目和检测结果。不过确实有一定参考意义作者: babybabe 时间: 2014-8-6 22:29
 图片附件: 57129036.snap.jpg (2014-8-6 22:30, 68 KB) / 该附件被下载次数 9
图片附件: 57129036.snap.jpg (2014-8-6 22:30, 68 KB) / 该附件被下载次数 9