标题:
【讨论帖】PCR问题交流
[打印本页]
作者:
@STAR@
时间:
2014-9-25 18:00
标题:
【讨论帖】PCR问题交流
各位站友
,多日以前,曾遇到一位朋友,让我来这和大家交流交流一些分子生物学的经验,一时太忙,落下了,今日在这开一贴,欢迎大家交流
作者:
baidukk
时间:
2014-9-25 18:00
不知群里是否有做多重PCR的兄弟姐妹,在这里想请教一下下面的多重PCR程序是如何设计出来的,需要依据什么来设计?请高人指点。
50℃ 2min
95℃ 10min
(95℃ 45s
68℃ 60s) 30cycles
(95℃ 30s
54℃ 30s
68℃ 60s)30cycles
68℃ 10min
作者:
2541
时间:
2014-9-25 18:01
我想请教一下聚合酶延伸效率的问题。一般的聚合酶的延伸效率是多少?在做pcr的时候,为什么20s就可以扩增出2K的片段?很迷惑。
作者:
redbutterfly
时间:
2014-9-25 18:01
如何pcr新基因?
作者:
@STAR@
时间:
2014-9-25 18:01
QUOTE:
原帖由
2541
于 2014-9-25 18:01 发表
bbcodeurl('http://bbs.antpedia.com/images/common/back.gif', '
%s
')
我想请教一下聚合酶延伸效率的问题。一般的聚合酶的延伸效率是多少?在做pcr的时候,为什么20s就可以扩增出2K的片段?很迷惑。
一般的来讲,Taq聚合酶的延伸速度是1000bp/min
至于你说在20s延伸出2000的片段这个有可能,但并不常见
一般在非特异扩增或者以逆转录产物做模板的时候会出现。
作者:
@STAR@
时间:
2014-9-25 18:03
QUOTE:
原帖由
redbutterfly
于 2014-9-25 18:01 发表
bbcodeurl('http://bbs.antpedia.com/images/common/back.gif', '
%s
')
如何pcr新基因?
你是指吊取未知基因是吗,这样的话,一般是根据这个未知基因在不同物种间的同源性在保守区设计同源简并引物进行PCR如果要获得全长基因的话还可能要在两端进行RACE扩增获得5'端和3'端非编码序列。
作者:
whitesheep
时间:
2014-9-25 18:04
我想请教一下:
我的目的片段是DNA长度5700多,CDNA长度1900,起初想从DNA中扩增目的条带,但是一直没有扩增出来,因此我提取RNA,合成CDNA第一链,这回有条带了,但是条带很弱,我的PCR条件:
1. 94° 3min
2. 94° 30sec
3. 51° 1min
-0.1°每个循环
5. 72° 3min
6. 2 ~5 32个循环
7. 72° 10min
我想进行胶回收然后,然后克隆该基因,不知道该如何优化a
作者:
2541
时间:
2014-9-25 18:04
先谢谢楼主,不过我扩出来的是目的条带,没有非特异条带。我正常情况下都按这个效率进行PCR的。
作者:
@STAR@
时间:
2014-9-25 18:05
我的目的片段是DNA长度5700多,CDNA长度1900,起初想从DNA中扩增目的条带,但是一直没有扩增出来,因此我提取RNA,合成CDNA第一链,这回有条带了,但是条带很弱,我的PCR条件:
1. 94° 3min
2. ...
================================
CDNA1900?另外你是用的长距离taq酶么?
作者:
2541
时间:
2014-9-25 18:05
长的和普通的都用了!
作者:
2541
时间:
2014-9-25 18:06
谢谢您的回答!还有就是我现在重新P,原来的条件竟然没有目的带了!
作者:
2541
时间:
2014-9-25 18:06
CDNA长度1900多!
作者:
wiwi
时间:
2014-9-25 18:06
1000bp/min,保险起见,可适当延长时间
作者:
@STAR@
时间:
2014-9-25 18:07
如果你是以逆转录产物做模板扩增cDNA的话,你的条件要改改,你的延伸时间2分钟足够,退火温度放到50度就可以
那个-0.1度那步不要,没啥用,你一旦扩增出目的条带,把它回收了之后,以产物做模板提高退火温度再P一次
作者:
TNT
时间:
2014-9-25 18:07
Taq酶1kb一分钟吧
作者:
dodoit
时间:
2014-9-25 18:08
请楼主帮忙回答一下这个问题,谢谢了
cuturl('http://ask.bbioo.com/ask/show-1922.html')
作者:
ENA
时间:
2014-9-25 18:08
各位 我在PCR时 没加模板 可总是有带,我一共有20多对不同的引物,不加模板,几乎都有带,不知道为什么!我的引物是分装的,而且 基本排除了污染。请问,这种情况,有无道理?
作者:
flower@@
时间:
2014-9-25 18:08
我也有个问题想请教,昨天做PCR,20微升体系,目的片段一个是300bp左右,一个是2300bp(、我当时不知道),我选的延伸时间是1min,结果做完电泳后发现后一个目的片段有两条很亮的条带,一个大约就是2000多,另一个在200~300之间,我用的酶是Pyrobest,高手请帮忙解释一下为什么会出现这样的两条带?
作者:
yhz1973
时间:
2014-9-25 18:08
请问一下,为何我的pcr产物检测时条带很浅,我已经设置了退火温度梯度,模板也是才提的,最近跑出的pcr条带一直很浅,请大家帮帮忙
作者:
chuntian1983
时间:
2014-9-25 18:09
各位 我在PCR时 没加模板 可总是有带,我一共有20多对不同的引物,不加模板,几乎都有带,不知道为什么!我的引物是分装的,而且 基本排除了污染。请问,这种情况,有无道理?
===========================================================================================================
我在做荧光定量PCR的时候也遇到过这种问题 后来觉得可能是由于荧光定量这种东西过于敏感 一点点模板就可能p出东西来 再有一点就是 可以试试把枪酒精消毒 再放到紫外下照 全面消毒 或者用带滤膜的枪头 有时候 枪和环境中一些残留的东西也会作为模板出现
作者:
wood533
时间:
2014-9-25 18:09
为什么我做单管PCR时可以P出来,而做96孔板却P不出来了呢?想了半天想不出原因,给指点下哪些方面应该特别注意。
作者:
@STAR@
时间:
2014-9-25 18:10
请楼主帮忙回答一下这个问题,谢谢了
cuturl('http://ask.bbioo.com/ask/show-1922.html')
===========================================================================================================
首先一点你要确定你的目的,如果你要获得该基因的全长,包括5’帽子3’尾巴的话你就要扩增全部的cds并做RACE获得末端序列。
如果你要做基因表达,那么你就要确定你表达哪一部分,如果是表达全长的话你就要设计引物扩增全部的CDS并在引物的5’端引入酶切位点,如果泥只需要扩增它的活性功能区,那么你就要找到CDS中的成熟肽序列并设计带酶切位点的引物扩增。
至于具体的设计方法,请参考论坛其他引物设计的帖子,具体问题具体分析。
作者:
@STAR@
时间:
2014-9-25 18:10
各位 我在PCR时 没加模板 可总是有带,我一共有20多对不同的引物,不加模板,几乎都有带,不知道为什么!我的引物是分装的,而且 基本排除了污染。请问,这种情况,有无道理?
===========================================================
没加模板有条带和你正常,那是因为形成了引物二聚体,一般很小,不超过200
作者:
@STAR@
时间:
2014-9-25 18:11
我也有个问题想请教,昨天做PCR,20微升体系,目的片段一个是300bp左右,一个是2300bp(、我当时不知道),我选的延伸时间是1min,结果做完电泳后发现后一个目的片段有两条很亮的条带,一个大约就是2000多,另一个在 ...
============================================
我没有弄明白你的问题,你是在一个体系里P两种产物吗?
作者:
@STAR@
时间:
2014-9-25 18:11
请问一下,为何我的pcr产物检测时条带很浅,我已经设置了退火温度梯度,模板也是才提的,最近跑出的pcr条带一直很浅,请大家帮帮忙
======================================================================================
这个有很多的原因,除了退火温度之外,跟你模板的浓度及质量,taq酶的质量,循环次数都有关系。
建议你在不改变其他条件的情况下,分别增加模板的浓度,降低退火温度如降至49-50度,增加Mg离子及taq酶浓度等等。
作者:
@STAR@
时间:
2014-9-26 09:49
为什么我做单管PCR时可以P出来,而做96孔板却P不出来了呢?想了半天想不出原因,给指点下哪些方面应该特别注意。
==================================================================================
一般采用大管配体系再分装至96孔板的方法,这样保证你的体系不至于出现问题,另外你期间改变其他条件了吗
作者:
whitesheep
时间:
2014-9-26 09:50
哇 延伸时间太长了吧 为什么要 -0.1°每个循环?不太有用吧!
作者:
xue258
时间:
2014-9-26 09:51
请问我想PCR一个长约20,000 bp的基因,怎么分开做?谢谢各位大虾!
作者:
qqq111
时间:
2014-9-26 09:51
前阵子,出差了,马上接着做,看看能不能出来!!谢谢各位达人帮忙!!!
作者:
glass
时间:
2014-9-26 09:51
请教高手们,有扩增出10kb以上的片段的吗?
作者:
XYZQ
时间:
2014-9-26 10:40
我想请问一下 我做了几组PCR但跑出来的条带相同的 包括亲本和子代 问题是 试剂没有污染
作者:
@STAR@
时间:
2014-9-26 10:54
请教高手们,有扩增出10kb以上的片段的吗?
========================================================================
有啊,扩增长片段的PCR叫做长距离PCR,一般是扩增基因组DNA用的,需要用到特殊的酶和体系
作者:
@STAR@
时间:
2014-9-26 10:55
我想请问一下 我做了几组PCR但跑出来的条带相同的 包括亲本和子代 问题是 试剂没有污染
=================================
很有可能是因为你的引物特异性不好造成的
作者:
cocacola
时间:
2014-9-26 10:59
我想问一下PCR对模板的量有什么要求呢?一般多少?是不是越多越好?还有PCR电泳点样时对点样量有要求么?我刚进实验室,很多量的问题让我很头疼
作者:
@STAR@
时间:
2014-9-26 15:04
我想问一下PCR对模板的量有什么要求呢?一般多少?是不是越多越好?还有PCR电泳点样时对点样量有要求么?我刚进实验室,很多量的问题让我很头疼
======================================================
我不知道你是否认真看过宝生物的Taq酶说明书,上面说模板的浓度为
人基因组DNA 0.1ug-1ug
大肠杆菌基因组DNA 10ng-100ng
质粒DNA 0.1-10ng
λDNA 0.5-5ng
点样没有具体要求,用伯乐系统大孔最多能上50微(0.4g琼脂糖/40ml)
小孔20微
作者:
wiwi
时间:
2014-9-26 16:16
我想问问我在扩增已知序列基因的CDS ,设计引物时搜索到引物扩增模板不能全部包含CDS区,该怎么办。
作者:
@STAR@
时间:
2014-9-26 16:16
QUOTE:
原帖由
wiwi
于 2014-9-26 16:16 发表
bbcodeurl('http://bbs.antpedia.com/images/common/back.gif', '
%s
')
我想问问我在扩增已知序列基因的CDS ,设计引物时搜索到引物扩增模板不能全部包含CDS区,该怎么办。
一看就知道你是用软件设计引物的,实际上真正的引物设计应该是自己设计为主,软件为辅,自己根据具体要求设计好后用软件检测才是上策,你遇到的问题是因为软件本身的缺陷造成的。你设计引物的时候按照大的原则自己大胆设计,再用软件检测。你扩增CDs引物就一定要包括全部的CDS
作者:
whitesheep
时间:
2014-9-26 16:17
我想请问一下出现引物二聚体的原因有哪些?
是不是P不出条带的原因很多,dNTP、Mg、引物、DNA都有可能影响结果!
我每次是做2个引物,60个DNA 但是换了一批新的引物就老是白板,或者条带不清楚,还有二聚体的情况!这和染胶有关系么?
作者:
mnstyle
时间:
2014-9-26 16:18
目前,有许多生物公司都推出了快带Taq酶,就可以实现快速PCR,其实这是通过牺牲PCR的准确率来实现的,也就是Taq酶识别DNA序列更加“模糊”,这样才能大大提高速度。
当然也有那种速度又快,而准确率也较高的产品,但也是高端产品,普通的快速Taq酶,建议大家用作检测之用吧。
作者:
mnstyle
时间:
2014-9-26 16:18
我认为这种情况一般是引物设计不合适造成的,建议你重新设计引物,增加引物的TM值,有个问题,你已知目的基因组序列了吗?基因组序列是由内含子和外显子组成,如果你根据CDNA序列设计的引物,有可能引物序列正好跨在内含子外显子交界处,又或是之间的内含子序列太长,再就是基因组序列中有复杂结构,需要你添加GC ENHANCER等,来促进复杂结构的解链。
另外,你说之后的实验发现条带不见了,有可能是你CDNA降解的问题,不知道你的RNA质量怎么样,反转录的效率又怎么样
作者:
mnstyle
时间:
2014-9-26 16:22
这个应该是二聚体的原因,基因上引物二聚体很容易形成,
作者:
mnstyle
时间:
2014-9-26 16:23
Taq酶的延伸速度本来就是1-2KB/min,只不过针对于广泛的模板来说,保险的计算方法是1KB/min,其实Taq酶针对不同复杂程度的模板,其速度也不尽相同,一些比较简单的模板的延伸速度就是比较快一些,很正常。
作者:
mnstyle
时间:
2014-9-26 16:24
使用LA Taq即可,不过,LA Taq没有末端加A功能
作者:
mnstyle
时间:
2014-9-26 16:25
P不出条带的原因有很多,有可能是模板降解,引物溶解不好,或是PCR条带不好,当然也与buffer等有很大的关系,检查一下是否污染,buffer和DNTP一定要融化完全再取用。
要解决二聚体的根本方法就是重新设计引物,因为引物是最直接的原因,你可以试着减少引物用量,跟染胶是没有关系的
作者:
dragonkilly
时间:
2014-9-26 16:26
大家好!
我想在目的片段上加上linker的基因片段,听说可以用PCR 的方法P出来,我试着设计好了加上linker基因的引物,但是不知道在进行PCR时具体是如何操作的!请大家不吝赐教,谢谢了
作者:
nn255
时间:
2014-9-26 16:26
做PCR时,我们一般改变退火温度,循环数,那PCR的变性、退火、延伸的时间又应该怎么设置比较合适呢,我曾经试过三个温度相同,但变性、退火、延伸的时间稍有不同,结果就不一样了。我困惑,PCR的条件这么多,我们要怎么去找出合适的条件让我们实验得到满意的结果呢?
作者:
glass
时间:
2014-9-26 16:27
各位高人,大家好!我刚开始做实验,有个问题很困惑,想请教一下:我在做从全血提取DNA,请生物公司设计合成引物后进行PCR,跑胶为什么有些能出现条带,有些不能,跑出的条带在目的条带范围内,也没有二聚体,跑不出的就什么都没有,不知道什么原因。谢谢!
作者:
mnstyle
时间:
2014-9-26 16:27
就是直接P就可以了,只要你的Linker不是太长就行,
如果加的Linker长一点的话,前几个循环先降一下退火温度,之后再恢复正常退火温度。
作者:
mnstyle
时间:
2014-9-26 16:27
一般来说是有一个时间范围的,像变性温度随着模板的不同而变化,如基因组或菌液就需要变性时间较长,退火时间也是根据模板和引物的不同而有差异,延伸时间要看目的条带的复杂性及Taq酶的延伸效率限制。
总之,就是与模板的长短及复杂度和目的条带所限。
作者:
mnstyle
时间:
2014-9-26 16:29
首先,生物公司合成的引物也不一定就百分百成功啊。
其次,你提取DNA的质量怎么样,不敢保证。
再次,PCR条件可能不合适,需要优化一下,包括PCR的体系,如果有复杂结构,或是高GC序列,就需要用特殊的buffer了。
作者:
kswl870
时间:
2014-9-26 16:30
我想问问如果杂带很多应该是提高退火温度么?那么为什么?谢谢
作者:
mnstyle
时间:
2014-9-26 16:30
杂志太多,肯定是引物结合位点太多,也就是特异性不高,那就需要提高退火温度,增强特异性。
你最好试一下梯度PCR
作者:
idea2011
时间:
2014-9-26 16:30
请问LM-PCR的原理和应用?谢谢!
作者:
shenkunjie
时间:
2014-9-26 16:31
1.ISPCR中fish技术 是什么?
2.实时荧光定量PCR中,SYBR荧光染料渗入到双链时发光吗???还是等到PCR延伸到有荧光染料的时候才发光呢?请解释PCR产物增加同荧光素成正比的原理?
3.RACE技术和反向PCR有什么区别?我已经知道一段基因序列想要测得两端序列应该用哪一技术?
作者:
shenkunjie
时间:
2014-9-26 16:39
我做的是“荧光定量法做5种常用管家基因3-磷酸甘油醛脱氢酶、β-肌动蛋白、18S核糖体RNA 、28S核糖体RNA、β - 2 -微球蛋白、酸性核糖体磷蛋白P0在不同月龄籽鹅的不同组织中表达的稳定性研究”。
学生想请教您:
1、我用的是SYBR Green I法,在荧光定量时是否可以选择2-△Ct进行相对定量?
2、如果能选择2-△Ct进行相对定量,如何使其其扩增效率为1?如何检测?应该注意哪些问题?
作者:
shenkunjie
时间:
2014-9-26 16:40
请问,引物Tm值计算的问题,公式中的单价离子浓度,仅指K+浓度吗?要不要把Mg2+浓度算成2个K+的浓度。另外,不同的软件算同一引物的Tm值差别很大,请问以你的经验,哪种软件算的相对准确。
作者:
shenkunjie
时间:
2014-9-26 16:42
有没有遇到过死活扩不出目的基因的情况,如何分析问题并解决?
作者:
shenkunjie
时间:
2014-9-26 16:42
上下游引物的GC含量不能相差太大,为什么?
用mix的话是不是就不用摸索条件了。
mix的优缺点有哪些?
作者:
shenkunjie
时间:
2014-9-26 16:43
上下游gc含量相差太大,会造成退火温度的不同,不利于你的扩增,同样,用mix你也得考虑到你的引物和模板的结合,设置适当的退火温度
作者:
TNT
时间:
2014-9-26 16:43
请问做microRNA怎样设计loop-stem引物啊?使用TaqMan探针
作者:
xue258
时间:
2014-9-26 16:45
想用self-priming的方法将两个片段连接起来,片段A500bp(上下游引物分别为a,b),片段B 1700bp(上下游引物分别为c,d)。两个片段分别扩增的时候效果非常好,单一条带,也很亮,片段A,B混合,加引物a,d再扩,结果没能连上。两个片段大约有25bp的重叠,试过多种退火温度都不行,用的是TAKARA LA taq,请问要如何优化条件?
作者:
wiwi
时间:
2014-9-26 16:47
我在做基因的定点突变。一般基因定点突变的引物长度较长,25-35bp。这样引物的Tm值相应上升。在通常的情况下,Tm值决定退火温度,所以不应过高。但是《分子克隆》讲定点突变的引物Tm值一定要在75度或之上,同时其退火温度在55度-65度。。。。我不明白。。。请高手帮忙解答其对Tm值的要求和退火温度与Tm值间的怪异
作者:
zwsyrt
时间:
2014-9-26 17:01
1.ISPCR中fish技术 是什么?
2.实时荧光定量PCR中,SYBR荧光染料渗入到双链时发光吗???还是等到PCR延伸到有荧光染料的时候才发光呢?请解释PCR产物增加同荧光素成正比的原理?
3. ...
===========================================================================================================
1 FISH(fluorescence in situ hybridization)技术是一种重要的非放射性原位杂交技术。它的基本原理是:如果被检测的染色体或DNA纤维切片上的靶DNA与所用的核酸探针是同源互补的,二者经变性-退火-复性,即可形成靶DNA与核酸探针的杂交体。将核酸探针的某一种核苷酸标记上报告分子如生物素、地高辛,可利用该报告分子与荧光素标记的特异亲和素之间的免疫化学反应,经荧光检测体系在镜下对待测DNA进行定性、定量或相对定位分析。
2 SYBR荧光染料特异性地掺入DNA双链后,发射荧光信号,而不掺入链中的SYBR染料分子不会发射任何荧光信号,从而保证荧光信号的增加与PCR产物的增加完全同步。
3 当然用RACE技术,关于两者的区别你可以再仔细看看课件中的PPT
作者:
XYZQ
时间:
2014-9-26 17:03
想问下为什么pcr拖带这么严重,从洞孔一直下去
作者:
seagate
时间:
2014-9-26 17:04
是smear现象吧。。。是不是地毯带? 这是PCR体系有问题,能导致smear的原因太多,我不一一说了,你百度一下就可以找到。或者换个词搜索——“弥散”
作者:
seagate
时间:
2014-9-26 17:06
我在做基因的定点突变。一般基因定点突变的引物长度较长,25-35bp。。。。请高手帮忙解答其对Tm值的要求和退火温度与Tm值间的怪异
暗夜捕猎兽
===========================================================================================================
我不是高手,也斗胆说两句。
你是在做定点突变,那么突变点应该是在引物中间吧,也就是两边各有十几个bp的和模板完全配对,要注意,此时计算TM值时,只能计算5'端完全配对的序列,对于退火效率不高的酶,这个影响是很大的。所以即使你的引物长度有二三十个碱基,GC+TA计算TM超过七八十度,退火温度也只能放低,否则退火时连引物和模板的特异性结合都搭不上去~
或者你也可以尝试升温PCR,前5-10循环退火温度放低,有了足够量的完全匹配模板之后再用TM-5度左右退火完成PCR。
作者:
idea2011
时间:
2014-9-26 17:06
请问下引物少一个碱基的后果是什么?
作者:
dodoit
时间:
2014-9-26 17:08
DNA提取的质量对PCR影响大吗?为什么我的转化子(酵母)提取的DNA有时能扩增出来,有时重新提取后又不能扩增出来呢,如果,有影响那是怎么影响的?我怎么做才能提到满足PCr要求的DNA,请高人指点,
作者:
TNT
时间:
2014-9-26 17:10
我想请问一下:PCR用内参的作用及原理?
作者:
niangao1980
时间:
2014-9-26 17:10
请教一下扩增3000bp DNA 用来测序 PCR试验条件怎么摸 用的是LA酶 谢谢大家
作者:
xue258
时间:
2014-9-26 17:11
我想问下,我用线粒体基因组全序列为模板,设计引物扩增它,用primer5.0设计时,总是得不到理想的引物,总是出现错配,,是否因为引物长度相对基因组来说是很小的,所以很难避免它和基因组的非目的片段结合的位点很多,只能靠优化反应程序来解决???另外有文献报道扩增线粒体全序列所用的模板甚至是总DNA,那设计的引物其不是更容易错配,????所以很困惑啊,很纠结啊,,也很想弄明白。。高人指点
作者:
XYZQ
时间:
2014-9-26 17:11
我想请教一下做荧光定量PCR时引物和探针应该先设计哪个?
是先设计引物还是先设计探针然后设计引物?
作者:
birdfish
时间:
2014-9-26 17:11
请教一下,9月初的时候用primerstart高保真酶对质粒进行环式PCR扩增,进而实现对目的片段特异位点突变,退火温度53°的时候有目的片段,由于酶用完了,订购同样的酶继续做,可是在同样的条件,同样的模板引物竟然扩增不出来,然后我在此基础上又上下做了两个温度,还是什么都没有,请问这种情况该怎么处理呢?
作者:
@STAR@
时间:
2014-9-26 17:12
请问下引物少一个碱基的后果是什么?
===================================================================================================
你能否详细说明问题,是3'少了一个碱基还是5’还是在中间?一般来说,引物的中间少了会引起移码突变,3’少了影响不大,5'视情况而定
作者:
@STAR@
时间:
2014-9-26 17:12
DNA提取的质量对PCR影响大吗?为什么我的转化子(酵母)提取的DNA有时能扩增出来,有时重新提取后又不能扩增出来呢,如果,有影响那是怎么影响的?我怎么做才能提到满足PCr要求的DNA,请高人指点,
===================================================================================
当然有影响,一般来说你的DNA提的质量(浓度,纯度)越高,你后续的PCR结果越好,反之就会影响你的结果
作者:
@STAR@
时间:
2014-9-26 17:15
我想请问一下:PCR用内参的作用及原理?
===========================================================================================================
内参一般是用于定量PCR中组织表达分析用作参照,它是选用一些看家基因,看家基因在不同组织中表达是恒定的,这样就可以作为参照计算比较目的基因在不同组织中表达的差异
作者:
@STAR@
时间:
2014-9-26 17:15
请教一下扩增3000bp DNA 用来测序 PCR试验条件怎么摸 用的是LA酶 谢谢大家
=====================================================================
PCR中主要调整的条件就是退火温度,你这也属于长距离PCR了,建议你做退火温度的梯度
作者:
@STAR@
时间:
2014-9-26 17:15
我想问下,我用线粒体基因组全序列为模板,设计引物扩增它,用primer5.0设计时,总是得不到理想的引物,总是出现错配,,是否因为引物长度相对基因组来说是很小的,所以很难避免它和基因组的非目的片段结合的位点很多 ...
===========================================================================================================
首先说明一点,用软件设计引物不是不可,,但是电脑程序是死的,它不可能适应千变万化的模板序列,设计引物还是自己设计,只要几个大的原则把握住就行,其他的不用考虑。
作者:
ALALA
时间:
2014-9-26 17:19
可能你的其他试剂有DNA污染
作者:
大大大山楂
时间:
2014-9-26 17:20
您好!向您请教一个问题:一般在分子生物学试验中,想了解一个基因的功能,都怎么做?确切的说,具体的步骤如何?由于这方面的知识点非常零碎,我实在是想不明白了!谢谢您!辛苦!
作者:
04906
时间:
2014-9-26 17:21
(1)请高手指点一下,17bp的引物跑PCR的时候能扩出条带来?在做多重PCR的时候,特异性怎么样?(2)设计兼并引物的时候给公司下订单的时候能直接下吗?例如引物序列为:atcgNtttatgac,那个N需要注明比例是多少吗?
作者:
pou
时间:
2014-9-26 17:21
请教:在做RT-PCR过程中,不同试验组别的标本中提出的总RNA浓度不一样,在下一步生成cDNA的过程中,加样时是否需要把总RNA的浓度调成一样,否则不能进行组间的比较?
作者:
00无名指00
时间:
2014-9-26 17:22
模板相同,多对Tm值相当的引物混合之后进行PCR,扩增的条带大小相同,在50ul体系中,保证引物的总量达到10pmol,为什么扩增30个循环之后没有条带啊?
作者:
49888
时间:
2014-9-26 17:22
最近做overlap pcr 同一批的东西 前面做三次都出了 第四次就没了 一周之内做的
后面把三个片段都新换了一遍 结果做了一个月 不管怎么调节基因比例 改变pcr条件
一直是在应该出现目的条带的位置之上涂抹 都快疯掉了
还有一个问题就是拿胶回收的overlap产物做模板(检验过是好的) 也是扩不出来 电泳现象和上面overlap一样 我做的是单链抗体(scFv)基因的组装
高人能指点一下不?
作者:
viviwang1987
时间:
2014-9-26 17:26
用SDS法提取DNA时忘加KAc了,不过倒是提出了DNA的,请问对于PCR有影响吗?
还有,我跑PCR时老没条带,请各位高手指点指点。不胜感激!
作者:
xue258
时间:
2014-9-26 17:28
PCR扩增1600bp片段,一个月前的结果很好 每次都出结果最近半个月 用相同的条件做 都没结果 跑胶以后所有的产物都在孔里很亮 就是跑不出孔 用百分之一的胶
用的延伸时间是3分钟 不可能扩出来那么大片段的请教各位大侠 这是哪儿出了问题
作者:
orangecake
时间:
2014-9-26 17:33
PCR产物跑电泳没有发现杂带,但测序结果发现有杂带,该如何处理啊?急啊,谢谢!
作者:
Ao7
时间:
2014-9-26 17:33
高手,降落pcr测序如何设定,我的用温普通的pcrtm为67可以p出来,为什么降落pcr末了65度15个循环反而不可以
作者:
uaubc
时间:
2014-9-26 17:35
请教一下啊,我是知道cDNA的序列2500bp,但是想知道内含子和调控序列,BLAST后目的片段可能是4500左右,我提总DNA做模板,也扩出了片段,也回收了。但是目的片段太长,不知道怎么确定是不是我要的片段。请指点一下,有什么可以容纳大片段的载体啊?谢谢
作者:
tianmei001
时间:
2014-9-26 17:38
体系重要,避免非特异性扩增
作者:
zwsyrt
时间:
2014-9-26 17:39
怎样鉴定这个菌是我所要的菌呢?如何用PCR方法做啊?
我做的是结核菌
作者:
ALALA
时间:
2014-9-26 17:39
Taq酶一般是800~2000bp/min 平均是1200bp/min 具体效率跟体系,PCR条件,及模板,引物有关,一般情况下按1000.1200bp/min计算延伸时间,短片段适当延长,一般最好不要少于30sec,长片段适当减短,若10k的3-5min足够了,2k的一般1min10sec到1min30sec就行
作者:
zhenxin
时间:
2014-9-26 17:39
最近反转录的过程中遇到一件积极郁闷的事,开始做反转录pcr可以拉出目的片段(目的条带2100bp),可是过了一段时间怎么都p不出来了,我重新提了RNA,重新换了试剂,重新稀释了引物,可是还是拉布出来,但同样条件做的内参(750bp)每次都能p出来,目的片段就是拉不出来,而且每次都在100bp附近有模糊的条带,我是先将RNA用oligodT逆转录成cDNA,然后再用逆转录的cDNA为模板跑PCR,结果老是在100bp附近有条模糊的带————
作者:
niangao1980
时间:
2014-9-26 17:40
你们谁有转基因方面的资料 分享下啊 我打算做工程菌啊
但是现在还是很迷惑 谢谢
作者:
youreyes
时间:
2014-9-26 17:41
我想请教大家,我们做PCR实验共做了16条引物,8条探针的。前六条都能出峰,但是探针7、8不知道怎么回事就是不出峰,其它条件和前面的都是一样的,换了2套引物和2套探针结果都是一样的。探针7、8就是不出峰。想问问大家可能是什么原因?、、
作者:
zwsyrt
时间:
2014-9-26 17:41
我想问一下 以cDNA为模板和以DNA为模板 用同一个引物PCR扩增时 退火温度一样吗
作者:
XYZQ
时间:
2014-9-26 17:41
我想问一下,延伸时间对pcr很重要吗?
作者:
#甜#
时间:
2014-9-26 17:43
有一个问题一直让我觉得很困惑:定量PCR和定性PCR之前的区别是什么?
作者:
yes4
时间:
2014-9-26 17:43
虚心请教 如何看blast验证引物的结果 主要看哪些指标,是看连线?E值?还是其他的? 谢谢了!
作者:
feima+
时间:
2014-9-26 17:43
我想请教高手,PCR扩增是,气溶胶污染怎么办?气溶胶的污染程度有多大?
作者:
pulala
时间:
2014-9-26 17:44
至于”定量PCR和定性PCR“,个人认为:定量为product数量的多少,定性为product是否有。
作者:
pulala
时间:
2014-9-26 17:45
难以避免,Pcr仪器和准备pcr最好于俩不同实验室。
作者:
89tongzijun
时间:
2014-9-26 17:46
我是大三的学生,现在自己在做项目,想用PCR来扩增一个已知的基因,引物设计来自文献,但是PCR结果一直不好,仅有的一次反应温度体系设错了(stage3- 72℃ 10min设成了25个循环,而stage2只有一个循环),反而出了目的条带,其他的正确条件反而没有东西。请问这是什么原因呢?多谢!~~
作者:
xue258
时间:
2014-9-26 17:46
我想请教一下有关miRNA的stem-loop引物设计及用此方法做miRNA定量的问题。在cDNA中加入正向引物和通用引物后为什么引物二聚体特别多?怎么避免
作者:
dodoit
时间:
2014-9-26 17:47
PCR引物设计时正向引物和cDNA是完全相同的,反向引物和cDNA是反相互补的。可是在用stem-loop实验定量miRNA时,两个引物都是和cDNA一样的啊,为什么?
作者:
baidukk
时间:
2014-9-26 17:48
我是个PCR菜鸟?很多基本问题都不知道,比如我从文献上直接找的引物序列,但是不知道大小,我怎么才能知道它的大小啊?各位前辈们?
作者:
xiaoxiaoniao
时间:
2014-9-26 17:49
请教:常规PCR没有结果怎么办
用别的DNA试验,有目的片段出现,能说明引物没有问题吧?
用另一对引物做,也有不清楚的目的片段,是不是我的DNA样品有问题啊?
该如何改进呢?我的DNA样品是污泥的总DNA。
谢谢各位有经验的前辈给点建议~~~谢谢~~非常感谢~~
作者:
ha111
时间:
2014-9-26 17:49
TM值与退火温度的确定?
作者:
dragonkilly
时间:
2014-9-26 17:50
我用同样的反应体系和反应程序做PCR,有时能p出来,有时不行,原因何在??是我的操作有问题吗??
作者:
avi317
时间:
2014-9-26 17:50
求教高保真PCR,为什么只有引物二聚体
作者:
kswl870
时间:
2014-9-26 17:50
退火温度常与引物的长度和碱基组成有直接相关关系,引物越长或GC含量较高,退火的温度可以高些,反之则低些。
退火温度越高,引物与模板(即扩增的DNA)结合的特异性越高,这可大大减少非特异性DNA片段的扩增。退火温度:Tm-55˚C
作者:
huifeng0516
时间:
2014-9-26 17:51
在体内,tag酶就差不多1kb/min,通过优化可以达到7oobp/min 以上,
作者:
huifeng0516
时间:
2014-9-26 17:51
如果这种类似的基因有被克隆出来的,就仔细对比寻找同源序列,设计简并引物,pcr,如果没有,可以找到它的表达产物,推出序列,然后设计引物…,实在没法了就只有全基因克隆了……
作者:
seagate
时间:
2014-9-26 17:51
我也做pcr不过老是出现非特异性条带,郁闷的是,用水也扩地出来,引物模板,pcr混合物全换了,都没用,还请各位指点啊
作者:
panda王
时间:
2014-9-26 17:52
你好!我也是做定点突变的,也刚开始设计引物做PCR,对Tm值计算只能计算5‘端完全配对的还不理解,能麻烦你在解释下吗?非常感激!
作者:
uuooii
时间:
2014-9-26 17:52
我刚开始做RT-PCR1个多月,开始两个星期比较顺利,很快就得到目的条带,送去测序结果也比较满意;最近2周遇到问题了:与之前相同的体系、条件居然做不出来了,以前可以做出来的模板也出不来,但内参出的来。
现在不知道是条件不稳定呢还是引物问题?
作者:
whitesheep
时间:
2014-9-26 17:53
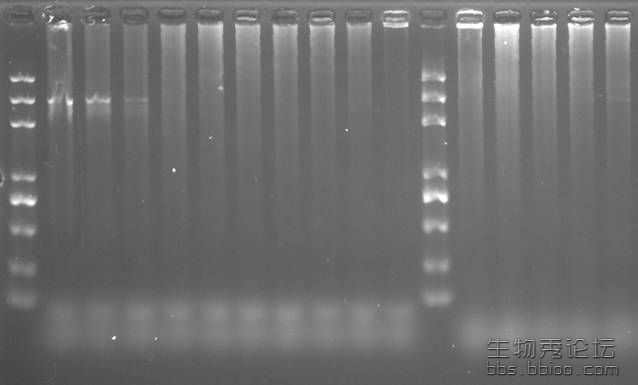
我扩增约3K的片段,以总DNA为模板,前几次都做了梯度,没做出来,于是重新抽屉总DNA,然后进行PCR扩增,从左到右是梯度,感觉前3个扩出来了但是不亮,但整体是一片拖的,可能是什么原因呢?
图是下面这样的情况
图片附件:
ͼƬ1.jpg
(2014-9-26 17:53, 17.15 KB) / 该附件被下载次数 10
http://bbs.antpedia.com/attachment.php?aid=27081
作者:
nut6694
时间:
2014-9-26 17:53
我有个师哥做实验 每次酶切成也成功了。也连接上了,就是扩增不出来,每次荧光检测就会出现就像墨滴印染的带 是什么原因呢?
作者:
idea2011
时间:
2014-9-26 17:53
请问ABI7000可以删除异常数据吗?
作者:
ENA
时间:
2014-9-26 17:54
大虾:请问如何解决mRNA抽提可能使低丰度蛋白丢失造成PCR扩不出来的问题啊?
作者:
IAM007
时间:
2014-9-26 17:54
新手求助,最近在摸索RT-PCR的条件,CT值在20-25之间,对数增长期的线谱还是很平顺的,但是到了平台期,线谱都有上下波动,线谱不平,请教各位,这有哪些原因造成,谢谢。
作者:
ritou1985
时间:
2014-9-26 17:54
如何设计引物?
作者:
vera+
时间:
2014-9-26 17:55
1000bp/min
作者:
vera+
时间:
2014-9-26 17:56
优化一下退火温度,要么就更改一下引物和模板的比例。
作者:
kswl870
时间:
2014-9-26 17:58
我想做SOE-PCR,请教引物如何设计?
作者:
@STAR@
时间:
2014-9-26 17:58
我做转基因植物PCR检测时,同样条件下阳性对照(目的基因质粒)条带很亮,但转化植株个别有目的带,但很不亮,转化植株模板DNA从1ul增加到3ul条带无明显变化,如何能让我的转化株条带亮起来
作者:
daod
时间:
2014-9-26 17:59
我做转基因植物PCR检测时,同样条件下阳性对照(目的基因质粒)条带很亮,但转化植株个别有目的带,但很不亮,转化植株模板DNA从1ul增加到3ul条带无明显变化,如何能让我的转化株条带亮起来
作者:
misswu61
时间:
2014-9-26 17:59
我做转基因植物PCR检测时,同样条件下阳性对照(目的基因质粒)条带很亮,但转化植株个别有目的带,但很不亮,转化植株模板DNA从1ul增加到3ul条带无明显变化,如何能让我的转化株条带亮起来
作者:
purrr
时间:
2014-9-26 17:59
pcr要大量做,预混的时候,加的试剂都有什么?什么不可以一起混?
作者:
vera+
时间:
2014-9-26 17:59
问个问题,
刚接触PCR技术,对于PCR试剂盒,其结果检测是否都需要电泳啊
作者:
junhun
时间:
2014-9-26 18:00
我的PCR做了好几回都没有跑出来,怎么判断PCR中是引物有问题?
作者:
白白的
时间:
2014-9-26 18:00
我想请教一下聚合酶延伸效率的问题。一般的聚合酶的延伸效率是多少?在做pcr的时候,为什么20s就可以扩增出 ...
=================================================================
看你用什么酶了 不同酶扩增速度不同 有一分1k的也有一分钟2k的啊 建议看看酶的说明书。
作者:
白白的
时间:
2014-9-26 18:01
我的PCR做了好几回都没有跑出来,怎么判断PCR中是引物有问题?
=============
让引物跑胶呗
作者:
喵咪
时间:
2014-9-26 18:01
我想请教一下聚合酶延伸效率的问题。一般的聚合酶的延伸效率是多少?在做pcr的时候,为什么20s就可以扩增出 ...
===========================================================================================================
其实是可以的,酶的活性是有一定的范围的,我记得我看过Taq酶有10s扩增1K的。像Phusion酶就是15——30s 1Kb的,说明给的都应该是比较保守的。
作者:
bgf5
时间:
2014-9-26 18:02
不知群里是否有做多重PCR的兄弟姐妹,在这里想请教一下下面的多重PCR程序是如何设计出来的,需要依据什么来设计?请高人指点。
50℃ 2min
95℃ 10min
(95℃ 45s
68℃ 60s) 30cycles
(95℃ 30s
54℃ 30s
68℃ 60s)30cycles
68℃ 10min
欢迎光临 分析测试百科 (http://bbs.antpedia.com/)
Powered by Discuz! 5.5.0
 图片附件: ͼƬ1.jpg (2014-9-26 17:53, 17.15 KB) / 该附件被下载次数 10
图片附件: ͼƬ1.jpg (2014-9-26 17:53, 17.15 KB) / 该附件被下载次数 10