正交试验法优选香砂养胃丸最佳打光工艺
 图片附件: 124004ebwjbv6zwtgpvptp.jpg (2015-3-12 11:00, 62.91 KB) / 该附件被下载次数 36
图片附件: 124004ebwjbv6zwtgpvptp.jpg (2015-3-12 11:00, 62.91 KB) / 该附件被下载次数 36 图片附件: 124032v0d67y7yg0mub7z7.jpg (2015-3-12 11:01, 45.55 KB) / 该附件被下载次数 33
图片附件: 124032v0d67y7yg0mub7z7.jpg (2015-3-12 11:01, 45.55 KB) / 该附件被下载次数 33
GMP认证前25项最后提醒
提升中药注射剂无菌保证水平
中药提取浓缩生产过程的自动控制
图片附件: 164717ejk42hlrptjveye8.jpg (2015-3-12 11:03, 32.26 KB) / 该附件被下载次数 34 图片附件: 164737qhsgs7xmdsf79txs.jpg (2015-3-12 11:03, 37.89 KB) / 该附件被下载次数 33
图片附件: 164737qhsgs7xmdsf79txs.jpg (2015-3-12 11:03, 37.89 KB) / 该附件被下载次数 33 图片附件: 164802abbxxz6bun2hvqq8.jpg (2015-3-12 11:04, 15.88 KB) / 该附件被下载次数 23
图片附件: 164802abbxxz6bun2hvqq8.jpg (2015-3-12 11:04, 15.88 KB) / 该附件被下载次数 23
消毒检测一次完成
无菌药品质量控制的注意要点
GMP洁净区干雾化过氧化氢空间灭菌系统简介
冻干工艺配制中的药液过滤
图片附件: 235929kwe339u233ww91or.gif (2015-3-12 14:28, 31.49 KB) / 该附件被下载次数 14
加强除菌过滤器的质量管理 有效降低注射剂类产品的无菌风险
冻干粉针工艺
粉针剂认证要点
浅淡注射器的压塞技术
图片附件: 235217sq0ohgsh900cu72h.gif (2015-3-12 14:31, 21.8 KB) / 该附件被下载次数 15 图片附件: 235240yj24676a25koj38h.gif (2015-3-12 14:31, 24.42 KB) / 该附件被下载次数 12
图片附件: 235240yj24676a25koj38h.gif (2015-3-12 14:31, 24.42 KB) / 该附件被下载次数 12 图片附件: 235324ua9fpj44irjd90qk.gif (2015-3-12 14:32, 32.62 KB) / 该附件被下载次数 11
图片附件: 235324ua9fpj44irjd90qk.gif (2015-3-12 14:32, 32.62 KB) / 该附件被下载次数 11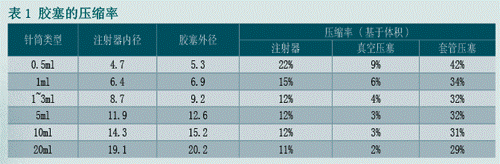
树立QbD理念以更好地保证产品的最终质量及用药安全
图片附件: 232406zmf3gukazuuqf5uo.jpg (2015-3-12 14:34, 22.23 KB) / 该附件被下载次数 11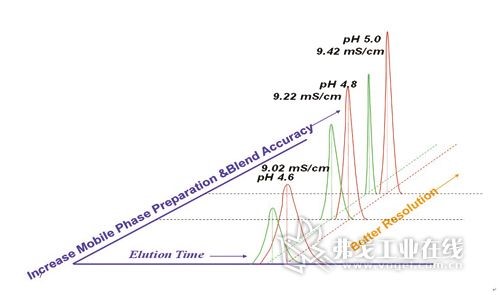 图片附件: 232425lk8s1qj9l97lzlc8.jpg (2015-3-12 14:35, 23.33 KB) / 该附件被下载次数 10
图片附件: 232425lk8s1qj9l97lzlc8.jpg (2015-3-12 14:35, 23.33 KB) / 该附件被下载次数 10 图片附件: 232438dlqnn3v7q5qzqxoo.jpg (2015-3-12 14:35, 34.29 KB) / 该附件被下载次数 80
图片附件: 232438dlqnn3v7q5qzqxoo.jpg (2015-3-12 14:35, 34.29 KB) / 该附件被下载次数 80 图片附件: 232504csbg7y7msalghtcg.jpg (2015-3-12 14:36, 18.68 KB) / 该附件被下载次数 12
图片附件: 232504csbg7y7msalghtcg.jpg (2015-3-12 14:36, 18.68 KB) / 该附件被下载次数 12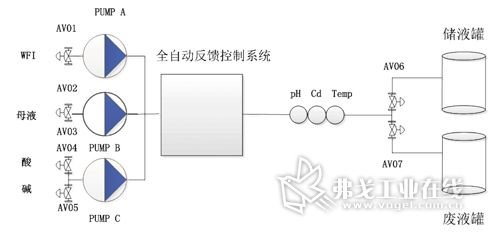 图片附件: 232524fxj8ntfxvxlrqmf1.jpg (2015-3-12 14:36, 44.5 KB) / 该附件被下载次数 9
图片附件: 232524fxj8ntfxvxlrqmf1.jpg (2015-3-12 14:36, 44.5 KB) / 该附件被下载次数 9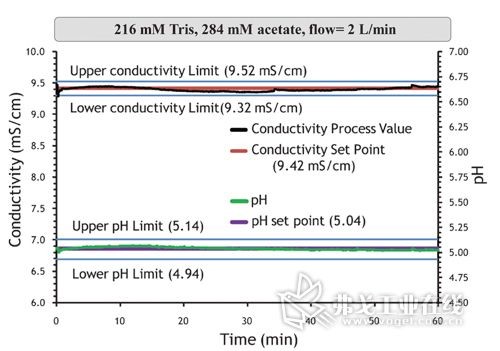
把风险降低到最小程度--避免无菌生产线的设计错误
图片附件: 140238e3t9iuez4mu39i9x.jpg (2015-3-12 14:37, 32.74 KB) / 该附件被下载次数 11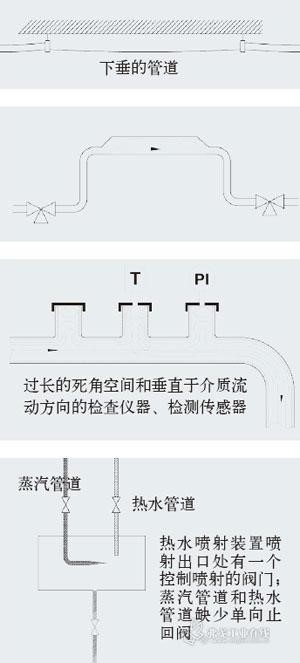
冻干过程的几个关键概念
制药级硅胶管的优点和局限性
图片附件: 121743p5yfwl7w8vs6990w.jpg (2015-3-12 14:39, 31.17 KB) / 该附件被下载次数 10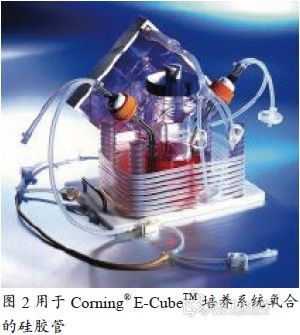 图片附件: 121809ypmum8pzk3vaen1d.jpg (2015-3-12 14:39, 19.78 KB) / 该附件被下载次数 5
图片附件: 121809ypmum8pzk3vaen1d.jpg (2015-3-12 14:39, 19.78 KB) / 该附件被下载次数 5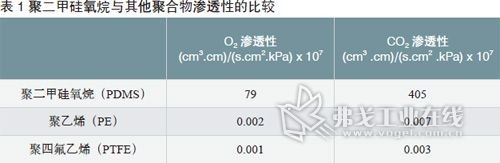 图片附件: 121838yj30k4pmop8qp81r.jpg (2015-3-12 14:39, 56.36 KB) / 该附件被下载次数 5
图片附件: 121838yj30k4pmop8qp81r.jpg (2015-3-12 14:39, 56.36 KB) / 该附件被下载次数 5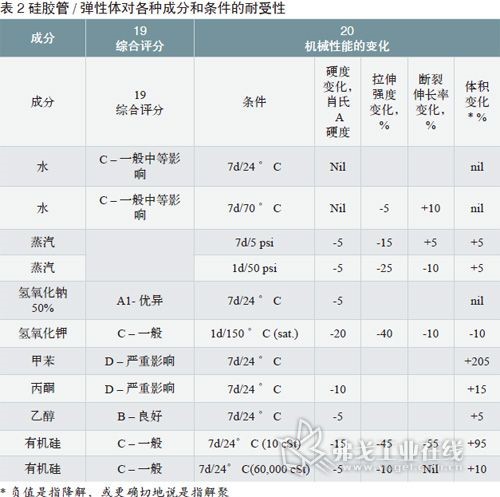 图片附件: 121904k9iw0kj04i4w6em2.jpg (2015-3-12 14:40, 17.99 KB) / 该附件被下载次数 7
图片附件: 121904k9iw0kj04i4w6em2.jpg (2015-3-12 14:40, 17.99 KB) / 该附件被下载次数 7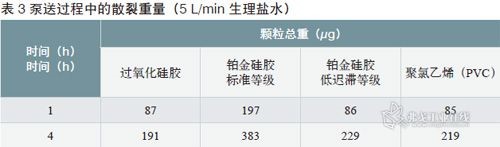
多级喷雾干燥塔的设备运行规则
喷雾干燥塔的能源节约
冻干设备应改进的问题解析
冻干技术在基因工程药物中的应用
冻干制剂经验谈
牛血清的作用与质量要求
细胞培养从头学
发酵工业杂菌污染的控制技术
新药(IND)除菌过滤的验证和确认
制粒技术
制粒过程的质量控制
制粒技术
真正实现无菌转移
图片附件: 132212atfanvi055itasi6.jpg (2015-3-12 15:02, 30.6 KB) / 该附件被下载次数 12 图片附件: 132308bjsrj35u3j5zok1j.jpg (2015-3-12 15:02, 13.93 KB) / 该附件被下载次数 17
图片附件: 132308bjsrj35u3j5zok1j.jpg (2015-3-12 15:02, 13.93 KB) / 该附件被下载次数 17
质量风险管理连接质量管理与精益生产
图片附件: 111702vqf9ig4i2tg0hgiq.jpg (2015-3-12 15:03, 10.93 KB) / 该附件被下载次数 16 图片附件: 111812xyryx56a30x0ss5l.jpg (2015-3-12 15:04, 30.41 KB) / 该附件被下载次数 12
图片附件: 111812xyryx56a30x0ss5l.jpg (2015-3-12 15:04, 30.41 KB) / 该附件被下载次数 12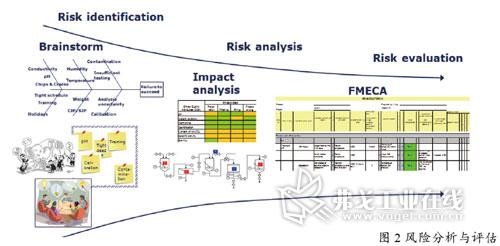 图片附件: 111837muwxg3aaf2vagjji.jpg (2015-3-12 15:04, 29.97 KB) / 该附件被下载次数 13
图片附件: 111837muwxg3aaf2vagjji.jpg (2015-3-12 15:04, 29.97 KB) / 该附件被下载次数 13
制药行业的干燥装置
切向流过滤技术的原理和应用
图片附件: 163914h262v2i41ljcl2lp.jpg (2015-3-12 15:09, 18.79 KB) / 该附件被下载次数 10 图片附件: 163959z1a6r0613x1va22z.jpg (2015-3-12 15:09, 41.7 KB) / 该附件被下载次数 7
图片附件: 163959z1a6r0613x1va22z.jpg (2015-3-12 15:09, 41.7 KB) / 该附件被下载次数 7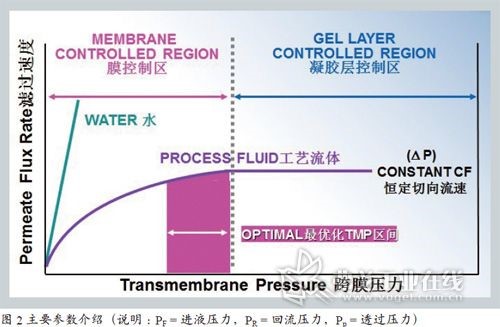 图片附件: 164025pcejdwhe4lh4h56k.jpg (2015-3-12 15:10, 27 KB) / 该附件被下载次数 9
图片附件: 164025pcejdwhe4lh4h56k.jpg (2015-3-12 15:10, 27 KB) / 该附件被下载次数 9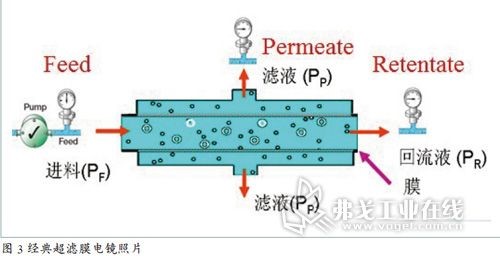 图片附件: 164044emgagga1dahl2p0a.jpg (2015-3-12 15:10, 27.3 KB) / 该附件被下载次数 11
图片附件: 164044emgagga1dahl2p0a.jpg (2015-3-12 15:10, 27.3 KB) / 该附件被下载次数 11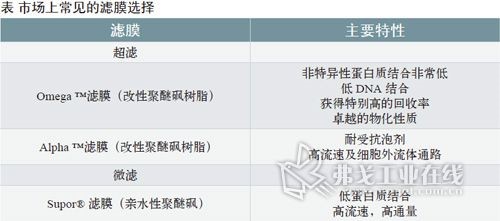
]湿热灭菌法
让洁净室工作更高效--洁净室连续监控系统设置和使用的几点建议
图片附件: 212201f3bjx0xzjeateozk.jpg (2015-3-12 15:16, 69.61 KB) / 该附件被下载次数 12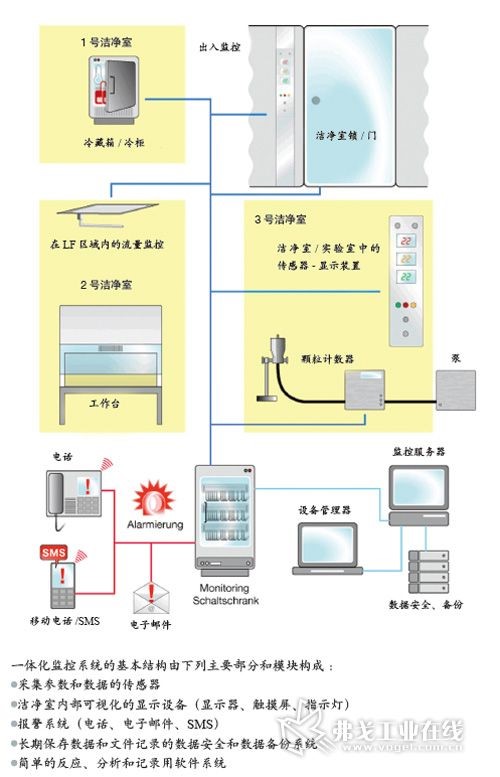
最大限度防控微生物污染
制药废水深度处理技术的研究
技术转移的差距分析6Ms运用
无菌生产工艺设计与风险控制
图片附件: 131908hrl4hhakkzlidj6z.jpg (2015-3-12 15:23, 28.29 KB) / 该附件被下载次数 14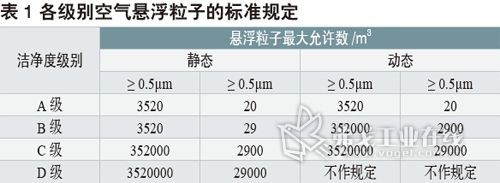 图片附件: 132029vo1b6eo1lcxs4bjc.jpg (2015-3-12 15:23, 25.25 KB) / 该附件被下载次数 17
图片附件: 132029vo1b6eo1lcxs4bjc.jpg (2015-3-12 15:23, 25.25 KB) / 该附件被下载次数 17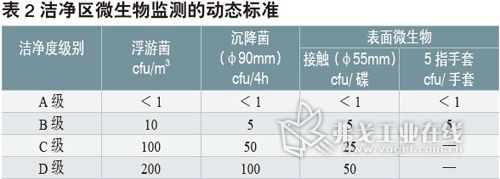 图片附件: 132057zugsrnguyblvzs8v.jpg (2015-3-12 15:24, 34.31 KB) / 该附件被下载次数 13
图片附件: 132057zugsrnguyblvzs8v.jpg (2015-3-12 15:24, 34.31 KB) / 该附件被下载次数 13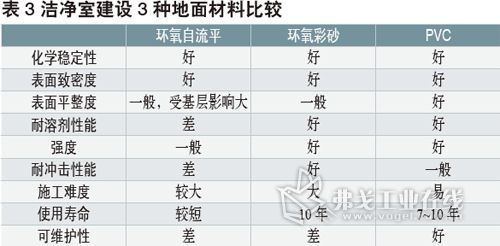
正确称重的实践建议
温湿度验证/分布研究的5个常见问题
无菌检查方法的验证
冻干工艺配制中的称量
冻干制剂经验谈
浅谈过氧化氢蒸汽灭菌与验证的相关问题
图片附件: 122142p6bjfhvchjcb7179.jpg (2015-3-12 15:33, 42.93 KB) / 该附件被下载次数 12 图片附件: 122218z8rlrf7pl8q7l77k.jpg (2015-3-12 15:33, 38.98 KB) / 该附件被下载次数 8
图片附件: 122218z8rlrf7pl8q7l77k.jpg (2015-3-12 15:33, 38.98 KB) / 该附件被下载次数 8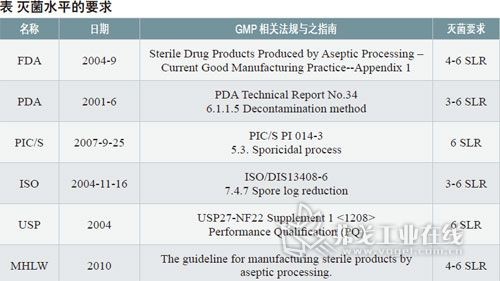
高效过滤器DOP检漏法在制药企业中的应用
探讨除菌级过滤器的重复使用(下)
图片附件: 024515d2gr27p3p7cpswa0.jpg (2015-3-12 15:37, 19.84 KB) / 该附件被下载次数 5 图片附件: 024627eksbngk08gnfcgxi.jpg (2015-3-12 15:37, 18.39 KB) / 该附件被下载次数 5
图片附件: 024627eksbngk08gnfcgxi.jpg (2015-3-12 15:37, 18.39 KB) / 该附件被下载次数 5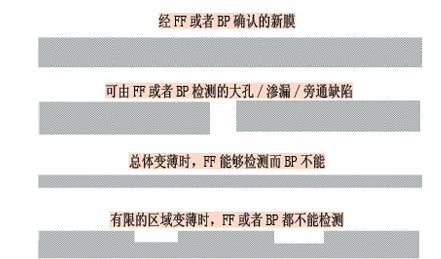
探讨除菌级过滤器的重复使用(上)
浅谈螺杆空压机的工作原理与流程
片剂生产总结(三)-包衣
片剂生产工艺总结(二)
片剂生产工艺总结(一)
解决中药注射剂澄明度的方法有哪些??
医药瓶设计中值得注重的几大因素
浅谈我国药用包装机械如何适GMP发展的需要
包装企业应怎样配备检测仪器?
针对目前常见药品包装质量问题的专家解答
国外瓶装片剂数颗机中通道式喂料器的观察与探讨
图片附件: 33868.jpg (2015-3-12 15:47, 14.86 KB) / 该附件被下载次数 8 图片附件: 33897.jpg (2015-3-12 15:47, 17.76 KB) / 该附件被下载次数 6
图片附件: 33897.jpg (2015-3-12 15:47, 17.76 KB) / 该附件被下载次数 6 图片附件: 33818.jpg (2015-3-12 15:47, 13.15 KB) / 该附件被下载次数 3
图片附件: 33818.jpg (2015-3-12 15:47, 13.15 KB) / 该附件被下载次数 3 图片附件: 34085.jpg (2015-3-12 15:48, 12.88 KB) / 该附件被下载次数 3
图片附件: 34085.jpg (2015-3-12 15:48, 12.88 KB) / 该附件被下载次数 3 图片附件: 34199.jpg (2015-3-12 15:48, 18.5 KB) / 该附件被下载次数 2
图片附件: 34199.jpg (2015-3-12 15:48, 18.5 KB) / 该附件被下载次数 2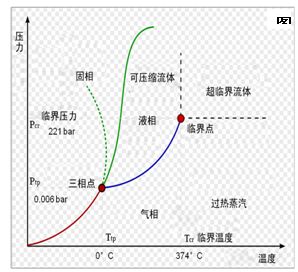 图片附件: 3134665.jpg (2015-3-12 15:49, 8.36 KB) / 该附件被下载次数 6
图片附件: 3134665.jpg (2015-3-12 15:49, 8.36 KB) / 该附件被下载次数 6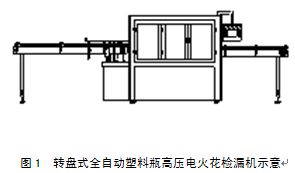
口服液洗瓶机所存在的问题
隧道式灭菌干燥机验证指标
新一代高速轧盖机研发思路及其特点
抗生素药物包装中丁基胶塞使用的有关问题
注射剂产品直接接触药品的包装材料和容器的选择考虑
直接接触的塑料包装材料指南(欧盟/欧洲药品管理局)
中药生产控制系统数据库二次开发
图片附件: 无标题.png.thumb.jpg (2015-3-12 16:06, 21.1 KB) / 该附件被下载次数 5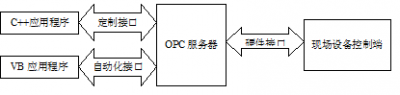 图片附件: 无标题.png.thumb (1).jpg (2015-3-12 16:07, 19.01 KB) / 该附件被下载次数 11
图片附件: 无标题.png.thumb (1).jpg (2015-3-12 16:07, 19.01 KB) / 该附件被下载次数 11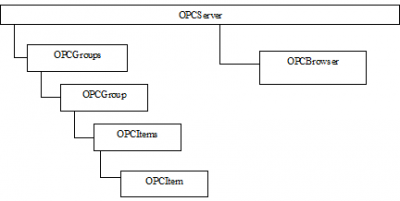 图片附件: 无标题.png.thumb (2).jpg (2015-3-12 16:07, 30.16 KB) / 该附件被下载次数 8
图片附件: 无标题.png.thumb (2).jpg (2015-3-12 16:07, 30.16 KB) / 该附件被下载次数 8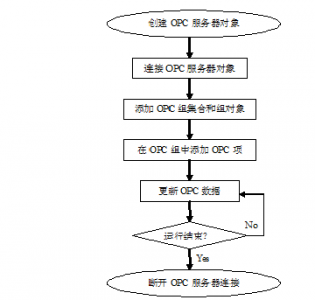 图片附件: 无标题.png.thumb (3).jpg (2015-3-12 16:08, 69.4 KB) / 该附件被下载次数 4
图片附件: 无标题.png.thumb (3).jpg (2015-3-12 16:08, 69.4 KB) / 该附件被下载次数 4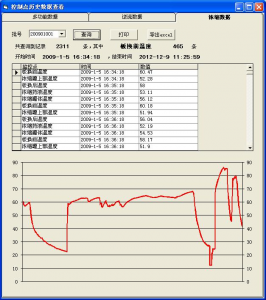
药厂微生物实验室的最佳实践
制剂如何通过TGA现场检查或认证
正确认识卫生型仪表及其合规性要求
湿法制粒小经验
抗生素玻璃瓶灌装加塞机的故障与排除
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |