选用固体制剂-瓶装包装生产线基本要求
药品颗粒数粒技术发展及应用
如何配置一条完美的瓶装包装线
铝塑泡罩包装机应用中的常见问题
]国产铝塑泡罩包装机与进口同类产品的对比分析
]简析药品电子监管码
 图片附件: 11293083.jpg (2015-3-12 16:28, 5.87 KB) / 该附件被下载次数 48
图片附件: 11293083.jpg (2015-3-12 16:28, 5.87 KB) / 该附件被下载次数 48
注射给药生产设备最新研究进展
图片附件: 11264241.jpg (2015-3-12 16:29, 15.33 KB) / 该附件被下载次数 48 图片附件: 11264586.jpg (2015-3-12 16:29, 9.4 KB) / 该附件被下载次数 49
图片附件: 11264586.jpg (2015-3-12 16:29, 9.4 KB) / 该附件被下载次数 49 图片附件: 11264628.jpg (2015-3-12 16:30, 12.56 KB) / 该附件被下载次数 23
图片附件: 11264628.jpg (2015-3-12 16:30, 12.56 KB) / 该附件被下载次数 23 图片附件: 11264666.jpg (2015-3-12 16:30, 10.64 KB) / 该附件被下载次数 33
图片附件: 11264666.jpg (2015-3-12 16:30, 10.64 KB) / 该附件被下载次数 33
药品包装与标签规范细则
真空包装机的分类和特点
包装技术和价格计算
盒类包装印刷实践的经验
包装覆膜工艺的分类
包装覆膜不亮原因解析
功能性纸质包装材料与纸箱
药品包装印刷的安全解决方法
双铝、铝铝 - 铝塑内包装材料的区别
浅谈药品包装工艺、材料与设备
应用缓冲系统优化包装效率
机器人技术在包装线上的应用分析
机器人技术提升生产效率
冷冻干燥设备性能选择以及配方冻干工艺验证
透明膜三维包装在制药中包装生产的应用
图片附件: 2009222201759319.gif (2015-3-12 16:42, 8.13 KB) / 该附件被下载次数 34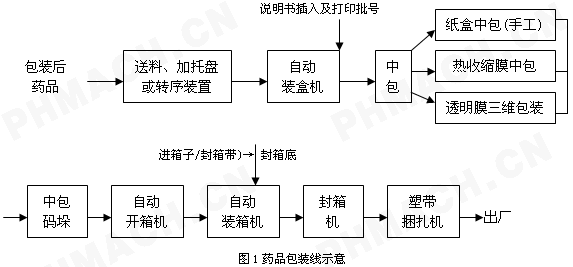
抗生素瓶洗瓶机的概要及选择要点
对瓶装联动线的基本组成与选择要点的探讨
图片附件: 2008124174215496.gif (2015-3-12 16:44, 22.4 KB) / 该附件被下载次数 32 图片附件: 2008124174320587.gif (2015-3-12 16:44, 11.13 KB) / 该附件被下载次数 30
图片附件: 2008124174320587.gif (2015-3-12 16:44, 11.13 KB) / 该附件被下载次数 30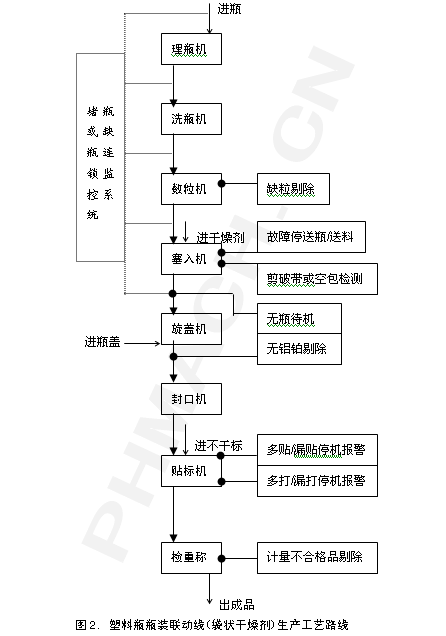
清洁验证的TOC方法--重新思考并审视清洁验证方法的适合度
过热水CIP和SIP系统为无菌原料药生产构建防线
GMP洁净区干雾化过氧化氢空间灭菌系统简介
生化药物生产过程中控制热原的方法
我国制药工业所用湿热灭菌设备状况分析
灭菌大法汇总
冻干机在线灭菌新工艺
过氧化氢蒸汽灭菌技术简介
关于过热水概念和制备
湿热灭菌
图片附件: 151531.jpg (2015-3-12 16:53, 19.04 KB) / 该附件被下载次数 29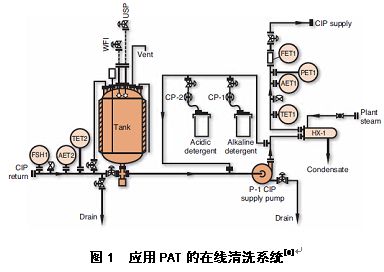 图片附件: 151549.jpg (2015-3-12 16:54, 10.38 KB) / 该附件被下载次数 21
图片附件: 151549.jpg (2015-3-12 16:54, 10.38 KB) / 该附件被下载次数 21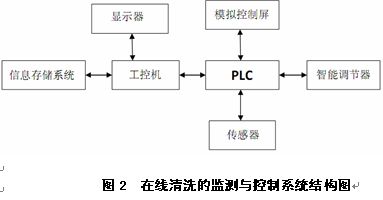
灭菌和无菌制剂及其制备技术发展概况
臭氧灭菌在药品生产环境中的应用
关于生物指示剂
超声波清洗及应用
湿热蒸汽灭菌柜的选用
灭菌
臭氧灭菌的残留物会对人体造成危害吗
对《无菌过滤系统在线灭菌与完整性测试的程序》的理解
图片附件: 200933072722.gif (2015-3-12 17:04, 6.09 KB) / 该附件被下载次数 32 图片附件: 2009330756963.gif (2015-3-12 17:05, 12.44 KB) / 该附件被下载次数 15
图片附件: 2009330756963.gif (2015-3-12 17:05, 12.44 KB) / 该附件被下载次数 15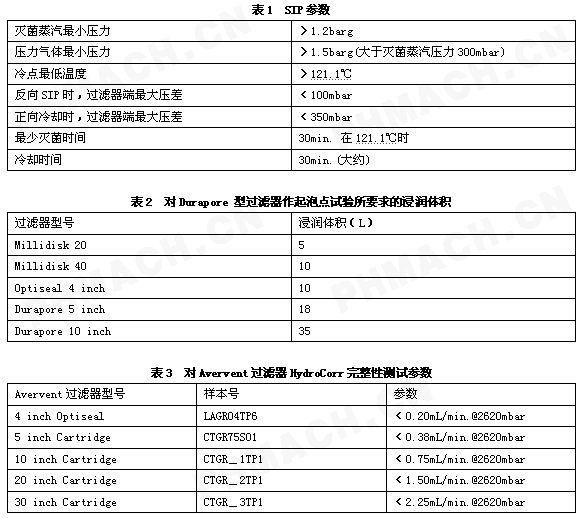 图片附件: 20093301014693.gif (2015-3-12 17:05, 11.93 KB) / 该附件被下载次数 17
图片附件: 20093301014693.gif (2015-3-12 17:05, 11.93 KB) / 该附件被下载次数 17 图片附件: 20093301039542.gif (2015-3-12 17:05, 7.05 KB) / 该附件被下载次数 29
图片附件: 20093301039542.gif (2015-3-12 17:05, 7.05 KB) / 该附件被下载次数 29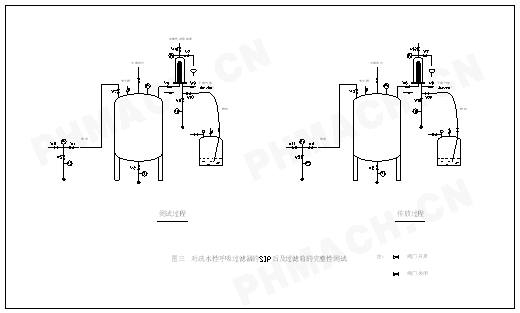 图片附件: 20093301234899.gif (2015-3-12 17:05, 5.47 KB) / 该附件被下载次数 32
图片附件: 20093301234899.gif (2015-3-12 17:05, 5.47 KB) / 该附件被下载次数 32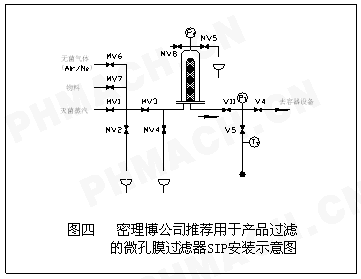 图片附件: 2009330170177.gif (2015-3-12 17:06, 5.97 KB) / 该附件被下载次数 23
图片附件: 2009330170177.gif (2015-3-12 17:06, 5.97 KB) / 该附件被下载次数 23 图片附件: 20093301941517.gif (2015-3-12 17:06, 6.83 KB) / 该附件被下载次数 17
图片附件: 20093301941517.gif (2015-3-12 17:06, 6.83 KB) / 该附件被下载次数 17
]对制药装备SIP命名与设计的探讨
超声波清洗设备使用方法
图片附件: paper_638_1.GIF (2015-3-12 17:09, 5.16 KB) / 该附件被下载次数 33 图片附件: paper_638_2.GIF (2015-3-12 17:09, 7.68 KB) / 该附件被下载次数 28
图片附件: paper_638_2.GIF (2015-3-12 17:09, 7.68 KB) / 该附件被下载次数 28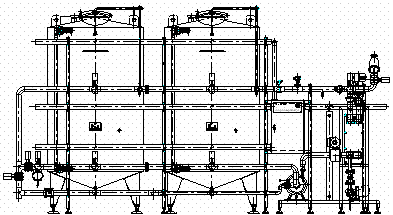
CIP系统的PLC控制系统设计
CIP系统打开清洁壁垒
制药设备SIP的科学性探讨
特殊药品存放区域、识别标志、贮存方法的规定
“运输”成药品冷链最大杀手
我国新修订药品GSP与欧盟原料药GDP之比较
重新加工、返工及回收几个易混淆概念释疑
实施2010版GMP困惑的思考
常用药品监管英语与缩略语

差压手工记录难以达到GMP要求
EU进口人用原料药的的问答(中英文)
MHRA的OOS指南与问答
中美GMP法规制度之对比分析
加拿大药监局GMP检查观察项风险分类
合理认识GMP的本质
药品生产中的变更控制
中国与欧美GMP的深层次差异
无菌产品欧美GMP检査员重点检査内容
欧盟食品接触材料指令解析
日本GMP法规制度简介
原料药开发与制造(Q11)
制剂如何通过TGA现场检查或认证
欧美药品GMP的主要特点
输液生产GMP考察团赴欧考察报告
ICH Q7A中的术语表
我国GMP与欧盟GMP的区别
对制药机械装备GMP验证的设想
以GMP理念提升制药装备水平
我国企业收到FDA警告信的分析
cGMP中文汇总
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |