欧盟CEP认证相关问题答疑
对FDA cGMP导言(Preamble)的理解
美国FDA的检查员制度值得我国借鉴
美国仿制药生物等效性评价的意义及方法
中美药品检查员队伍情况对比
FDA警告信制度
参加美国FDA2011工艺验证指南解读讲座总结
FDA-已批准申请的新药变更指南(中英文)
FDA有关术语大全
]CFR210/211原文件及中文翻译
FDA关于创新药物PAT过程分析技术的指导文件
 图片附件: 1114149.jpg (2015-3-14 10:17, 10.38 KB) / 该附件被下载次数 10
图片附件: 1114149.jpg (2015-3-14 10:17, 10.38 KB) / 该附件被下载次数 10
有关COS和EDMF知识
中国原料药DMF申报现状
美国DMF文件模板
]DMF(Drug Master File,药物主文件)怎么做?
美国FDA对企业的检查重点
特殊审批程序的沟通交流要点剖析
译稿:Process Validation–Key Areas Leading
关于21 CFR Part 11
FDA推行ICH-Q10
中国医药产品为何被美国FDA拒绝
中药通过FDA认证批准的基本要求
原料与医药中间体的FDA认证
FDA、欧盟所关注的重点验证内容
FDA认证流程
非PVC膜软袋大输液生产线正常生产中常见问题和解决办法
大输液玻璃瓶包装市场状况及发展浅析
药品的包装作用及要求
浅淡注射器的压塞技术
图片附件: 114145v8kik5zuctk4fluk.gif (2015-3-14 10:37, 23.99 KB) / 该附件被下载次数 26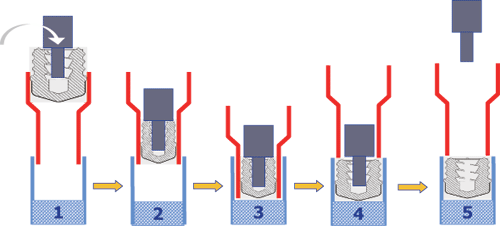 图片附件: 114208qhuwxh8aa7is4gad.gif (2015-3-14 10:38, 26.18 KB) / 该附件被下载次数 16
图片附件: 114208qhuwxh8aa7is4gad.gif (2015-3-14 10:38, 26.18 KB) / 该附件被下载次数 16 图片附件: 114228uucx7rqevuezo3xg.gif (2015-3-14 10:38, 22.08 KB) / 该附件被下载次数 24
图片附件: 114228uucx7rqevuezo3xg.gif (2015-3-14 10:38, 22.08 KB) / 该附件被下载次数 24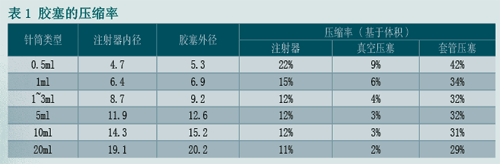
]玻璃预灌封注射器相关供应商评述及市场发展分析
预灌封注射器在中国之路
浅谈药品生产中电子监管码的实施方案
大输液生产过程中进行在线检漏的必要性
制药企业应合理选购中小型颗粒包装机
装盒机于制药工业中存在的问题及发展
全密闭类输液产品引领输液行业风潮
三合一无菌灌装 注射剂安全灌装
图片附件: 8152680.jpg (2015-3-14 10:46, 16.98 KB) / 该附件被下载次数 18 图片附件: 8152616.jpg (2015-3-14 10:46, 13.1 KB) / 该附件被下载次数 23
图片附件: 8152616.jpg (2015-3-14 10:46, 13.1 KB) / 该附件被下载次数 23
邦纳视觉产品在制药行业的应用
图片附件: 930465.gif (2015-3-14 10:50, 34.44 KB) / 该附件被下载次数 11 图片附件: 930439.jpg (2015-3-14 10:50, 58.43 KB) / 该附件被下载次数 16
图片附件: 930439.jpg (2015-3-14 10:50, 58.43 KB) / 该附件被下载次数 16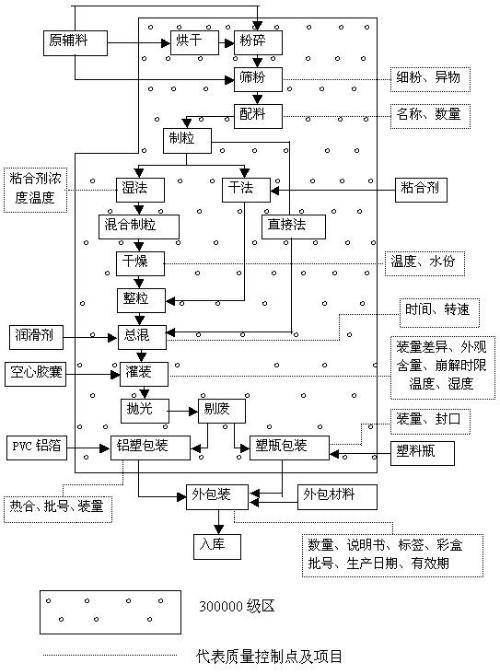 图片附件: 930435.jpg (2015-3-14 10:50, 14.99 KB) / 该附件被下载次数 14
图片附件: 930435.jpg (2015-3-14 10:50, 14.99 KB) / 该附件被下载次数 14 图片附件: 930424.jpg (2015-3-14 10:51, 15.5 KB) / 该附件被下载次数 7
图片附件: 930424.jpg (2015-3-14 10:51, 15.5 KB) / 该附件被下载次数 7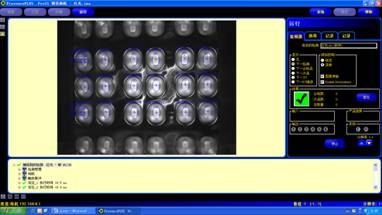 图片附件: 930464.jpg (2015-3-14 10:51, 11.04 KB) / 该附件被下载次数 10
图片附件: 930464.jpg (2015-3-14 10:51, 11.04 KB) / 该附件被下载次数 10 图片附件: 930499.jpg (2015-3-14 10:52, 10.44 KB) / 该附件被下载次数 12
图片附件: 930499.jpg (2015-3-14 10:52, 10.44 KB) / 该附件被下载次数 12 图片附件: 930585.jpg (2015-3-14 10:52, 17.13 KB) / 该附件被下载次数 6
图片附件: 930585.jpg (2015-3-14 10:52, 17.13 KB) / 该附件被下载次数 6 图片附件: 930516.jpg (2015-3-14 10:52, 17.96 KB) / 该附件被下载次数 17
图片附件: 930516.jpg (2015-3-14 10:52, 17.96 KB) / 该附件被下载次数 17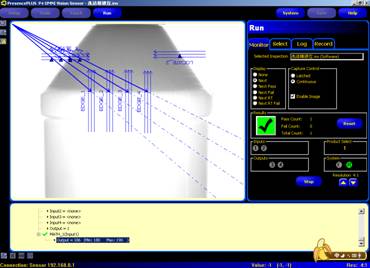 图片附件: 930526.gif (2015-3-14 10:54, 47.38 KB) / 该附件被下载次数 11
图片附件: 930526.gif (2015-3-14 10:54, 47.38 KB) / 该附件被下载次数 11 图片附件: 930589.jpg (2015-3-14 10:54, 13.22 KB) / 该附件被下载次数 12
图片附件: 930589.jpg (2015-3-14 10:54, 13.22 KB) / 该附件被下载次数 12 图片附件: 930525.jpg (2015-3-14 10:55, 19.81 KB) / 该附件被下载次数 15
图片附件: 930525.jpg (2015-3-14 10:55, 19.81 KB) / 该附件被下载次数 15 图片附件: 930538.gif (2015-3-14 10:55, 46.39 KB) / 该附件被下载次数 10
图片附件: 930538.gif (2015-3-14 10:55, 46.39 KB) / 该附件被下载次数 10 图片附件: 930572.jpg (2015-3-14 10:56, 28.92 KB) / 该附件被下载次数 13
图片附件: 930572.jpg (2015-3-14 10:56, 28.92 KB) / 该附件被下载次数 13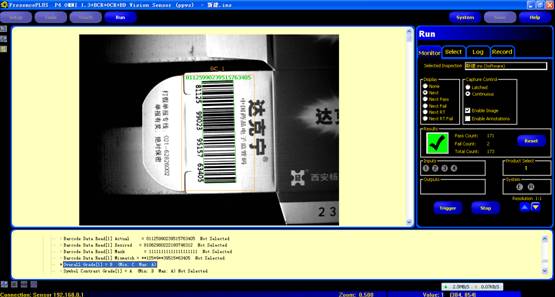 图片附件: 930655.gif (2015-3-14 10:56, 30.52 KB) / 该附件被下载次数 9
图片附件: 930655.gif (2015-3-14 10:56, 30.52 KB) / 该附件被下载次数 9 图片附件: 930620.jpg (2015-3-14 10:57, 17.02 KB) / 该附件被下载次数 5
图片附件: 930620.jpg (2015-3-14 10:57, 17.02 KB) / 该附件被下载次数 5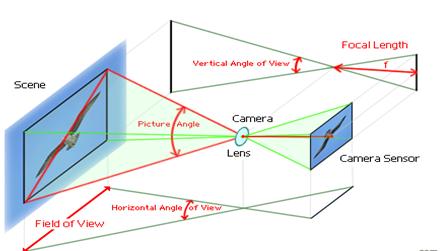 图片附件: 930662.gif (2015-3-14 10:58, 20.36 KB) / 该附件被下载次数 5
图片附件: 930662.gif (2015-3-14 10:58, 20.36 KB) / 该附件被下载次数 5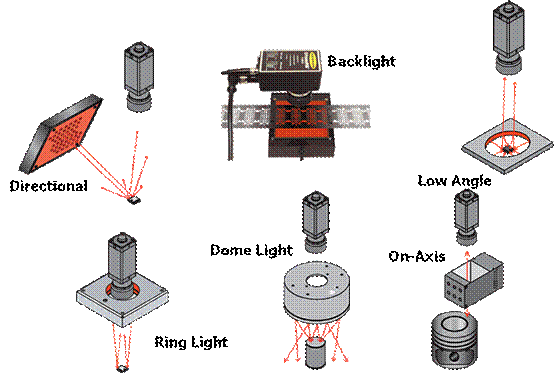
]国内外对药品包装安全的要求
图片附件: 5283810.jpg (2015-3-14 11:00, 35.25 KB) / 该附件被下载次数 11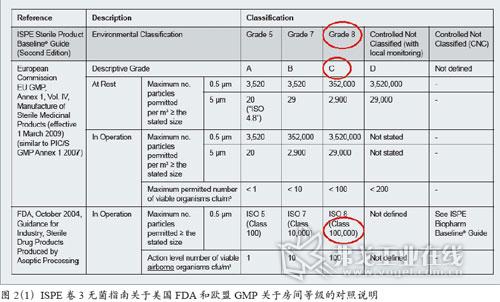 图片附件: 5283863.jpg (2015-3-14 11:00, 13.63 KB) / 该附件被下载次数 7
图片附件: 5283863.jpg (2015-3-14 11:00, 13.63 KB) / 该附件被下载次数 7 图片附件: 5283880.jpg (2015-3-14 11:00, 49.28 KB) / 该附件被下载次数 14
图片附件: 5283880.jpg (2015-3-14 11:00, 49.28 KB) / 该附件被下载次数 14 图片附件: 5283842.jpg (2015-3-14 11:06, 46.23 KB) / 该附件被下载次数 7
图片附件: 5283842.jpg (2015-3-14 11:06, 46.23 KB) / 该附件被下载次数 7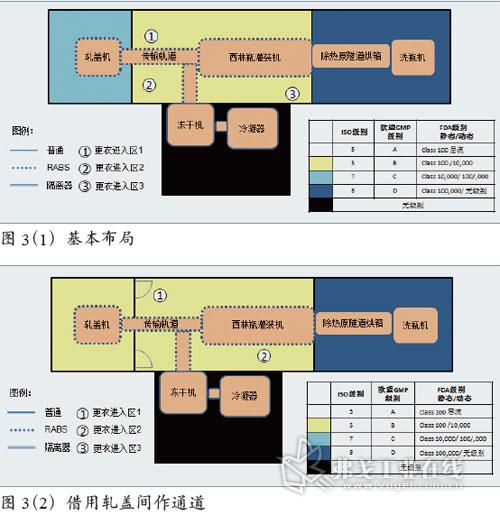 图片附件: 5283927.jpg (2015-3-14 11:07, 46.14 KB) / 该附件被下载次数 6
图片附件: 5283927.jpg (2015-3-14 11:07, 46.14 KB) / 该附件被下载次数 6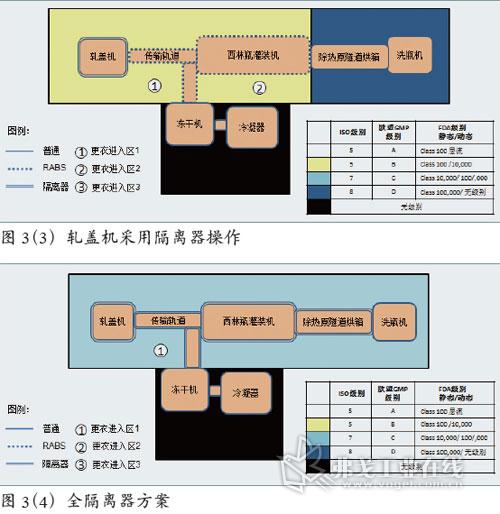 图片附件: 5283996.jpg (2015-3-14 11:10, 47.39 KB) / 该附件被下载次数 5
图片附件: 5283996.jpg (2015-3-14 11:10, 47.39 KB) / 该附件被下载次数 5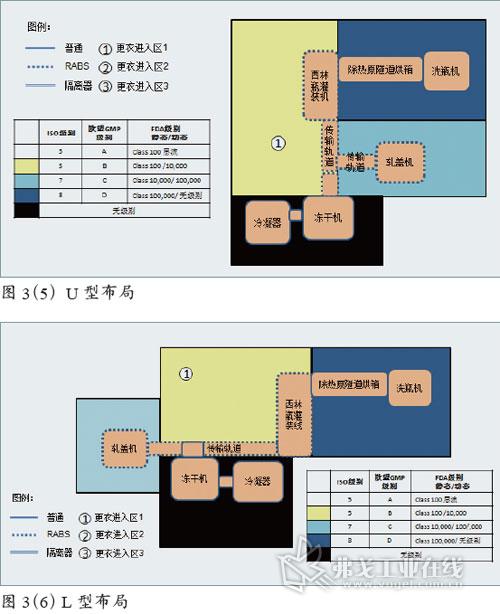 图片附件: 5283991.jpg (2015-3-14 11:11, 53.29 KB) / 该附件被下载次数 9
图片附件: 5283991.jpg (2015-3-14 11:11, 53.29 KB) / 该附件被下载次数 9 图片附件: 5283947.jpg (2015-3-14 11:13, 56.33 KB) / 该附件被下载次数 11
图片附件: 5283947.jpg (2015-3-14 11:13, 56.33 KB) / 该附件被下载次数 11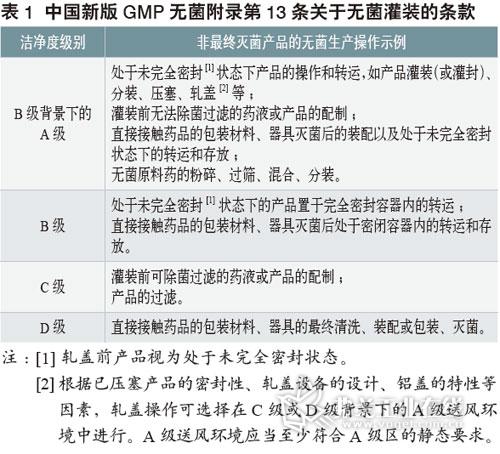
精益粉体加工,通往成功的钥匙
图片附件: 10235986.jpg (2015-3-14 11:16, 17.83 KB) / 该附件被下载次数 7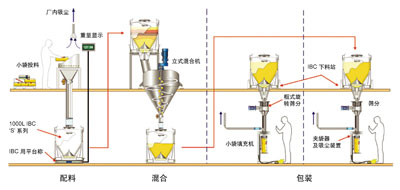 图片附件: 10235917.jpg (2015-3-14 11:17, 39.48 KB) / 该附件被下载次数 9
图片附件: 10235917.jpg (2015-3-14 11:17, 39.48 KB) / 该附件被下载次数 9 图片附件: 1023060.jpg (2015-3-14 11:18, 32.81 KB) / 该附件被下载次数 9
图片附件: 1023060.jpg (2015-3-14 11:18, 32.81 KB) / 该附件被下载次数 9 图片附件: 1023012.jpg (2015-3-14 14:41, 24.09 KB) / 该附件被下载次数 6
图片附件: 1023012.jpg (2015-3-14 14:41, 24.09 KB) / 该附件被下载次数 6 图片附件: 1023012.jpg (2015-3-14 14:41, 24.09 KB) / 该附件被下载次数 8
图片附件: 1023012.jpg (2015-3-14 14:41, 24.09 KB) / 该附件被下载次数 8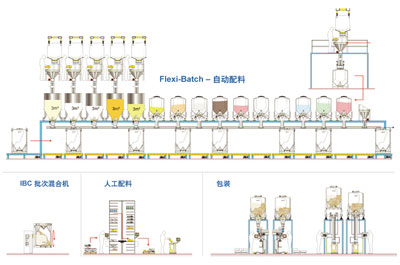 图片附件: 511446.jpg (2015-3-14 14:45, 15.96 KB) / 该附件被下载次数 8
图片附件: 511446.jpg (2015-3-14 14:45, 15.96 KB) / 该附件被下载次数 8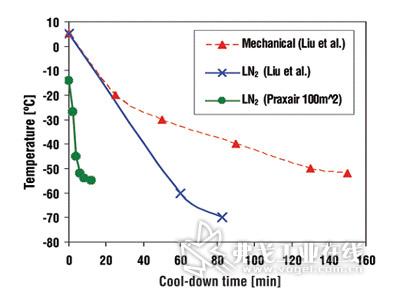 图片附件: 511438.jpg (2015-3-14 14:46, 23.05 KB) / 该附件被下载次数 10
图片附件: 511438.jpg (2015-3-14 14:46, 23.05 KB) / 该附件被下载次数 10
中国软包装输液的现状和未来发展趋势
无菌原料药生产中无菌工艺验证探讨
温度分布验证的8个步骤
对PDA发布的《工艺验证:一个生命周期方式》的解析
水针配制系统及药液输送管路清洁验证
******************有限责任公司
在GAMP5环境下的小型实验室设备的风险分析
图片附件: 1275372861.jpg (2015-3-14 15:08, 40.69 KB) / 该附件被下载次数 10 图片附件: 1275373267.jpg (2015-3-14 15:08, 54.6 KB) / 该附件被下载次数 6
图片附件: 1275373267.jpg (2015-3-14 15:08, 54.6 KB) / 该附件被下载次数 6 图片附件: 1275373199.jpg (2015-3-14 15:09, 226.57 KB) / 该附件被下载次数 5
图片附件: 1275373199.jpg (2015-3-14 15:09, 226.57 KB) / 该附件被下载次数 5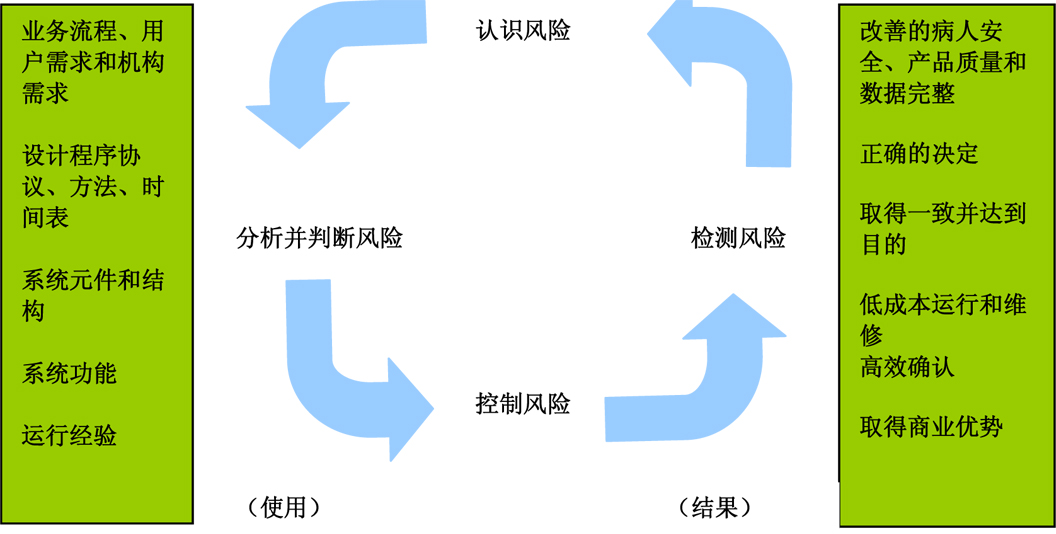 图片附件: 1275373331.jpg (2015-3-14 15:09, 186.65 KB) / 该附件被下载次数 6
图片附件: 1275373331.jpg (2015-3-14 15:09, 186.65 KB) / 该附件被下载次数 6 图片附件: 1275373277.jpg (2015-3-14 15:09, 222.06 KB) / 该附件被下载次数 6
图片附件: 1275373277.jpg (2015-3-14 15:09, 222.06 KB) / 该附件被下载次数 6 图片附件: 1275373106.jpg (2015-3-14 15:10, 309.79 KB) / 该附件被下载次数 8
图片附件: 1275373106.jpg (2015-3-14 15:10, 309.79 KB) / 该附件被下载次数 8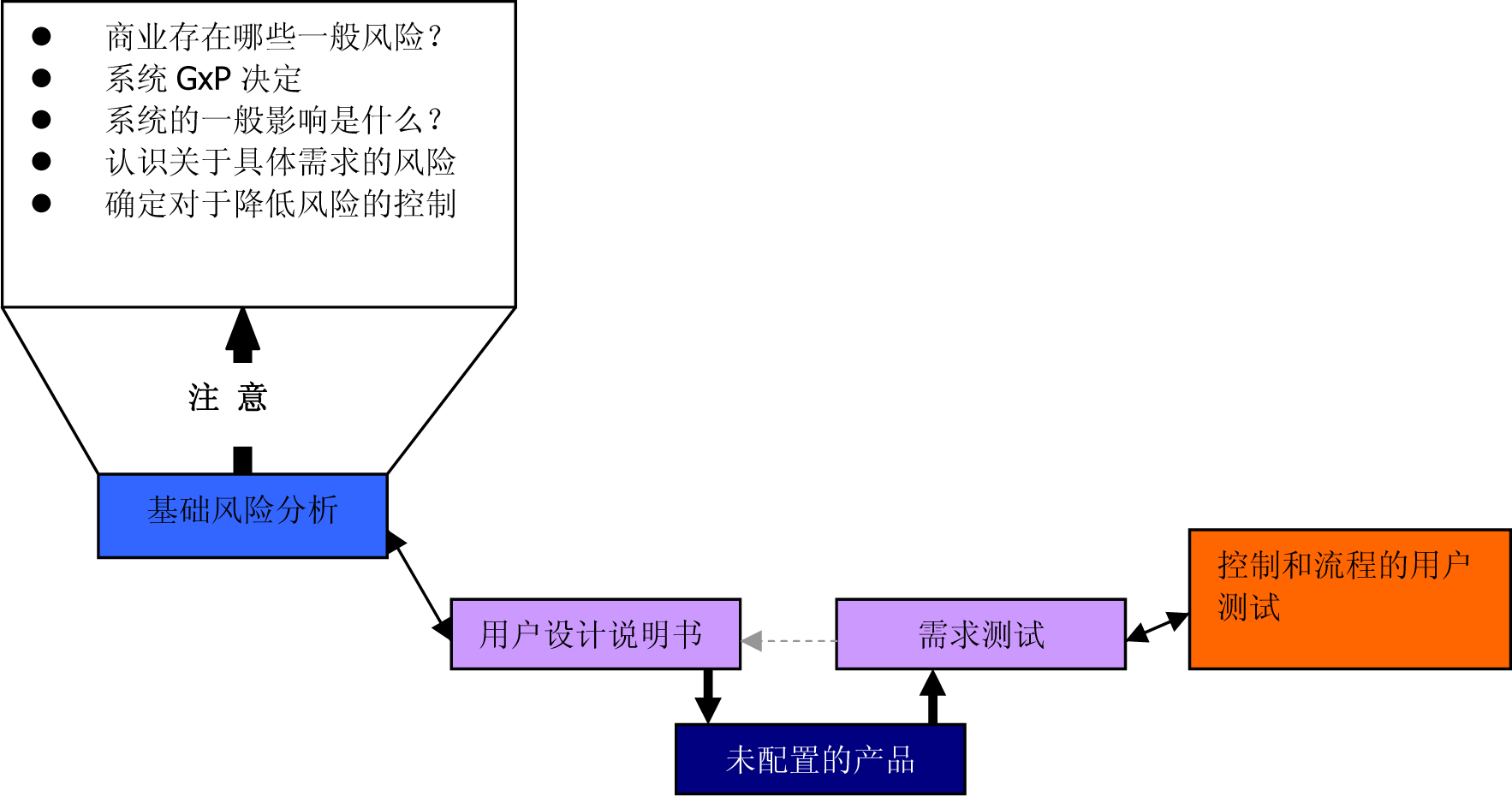
确认与验证:1℃精确挽救几个百分点的损失
]检验方法的验证与再验证管理(样文)
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |