验证总计划指南中文版
美国FDA清洗验证检查指南
FDA验证指导
USP无菌检验--隔离器系统验证
药品制备工艺研究和工艺验证的再次认识
隧道烘箱及干热烤箱除内毒素效果验证方法
在线总有机碳分析应用于清洁验证的可行性研究
 图片附件: 384096.jpg (2015-3-14 15:53, 12.16 KB) / 该附件被下载次数 6
图片附件: 384096.jpg (2015-3-14 15:53, 12.16 KB) / 该附件被下载次数 6 图片附件: 384086.jpg (2015-3-14 15:53, 33.74 KB) / 该附件被下载次数 4
图片附件: 384086.jpg (2015-3-14 15:53, 33.74 KB) / 该附件被下载次数 4 图片附件: 384026.jpg (2015-3-14 15:54, 14.97 KB) / 该附件被下载次数 5
图片附件: 384026.jpg (2015-3-14 15:54, 14.97 KB) / 该附件被下载次数 5
灭菌设备的验证方法及结果处理
风险评估技术在无菌粉针剂工程中的应用
小容量注射剂药液配制系统在线清洗验证
手提式不锈钢蒸汽消毒器的验证
图片附件: 185378.jpg (2015-3-14 16:47, 11.23 KB) / 该附件被下载次数 11
脉动真空灭菌器的常见问题及设备验证
图片附件: 16692.jpg (2015-3-14 17:00, 12.93 KB) / 该附件被下载次数 10 图片附件: 16656.jpg (2015-3-14 17:00, 11.16 KB) / 该附件被下载次数 8
图片附件: 16656.jpg (2015-3-14 17:00, 11.16 KB) / 该附件被下载次数 8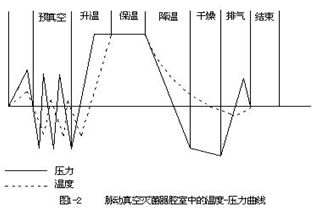 图片附件: 111235.jpg (2015-3-14 17:02, 55.79 KB) / 该附件被下载次数 12
图片附件: 111235.jpg (2015-3-14 17:02, 55.79 KB) / 该附件被下载次数 12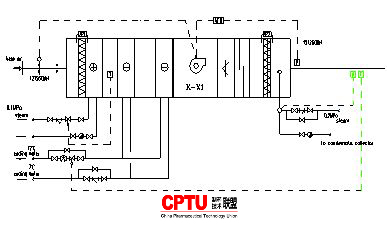 图片附件: 111346.jpg (2015-3-14 17:02, 77.46 KB) / 该附件被下载次数 14
图片附件: 111346.jpg (2015-3-14 17:02, 77.46 KB) / 该附件被下载次数 14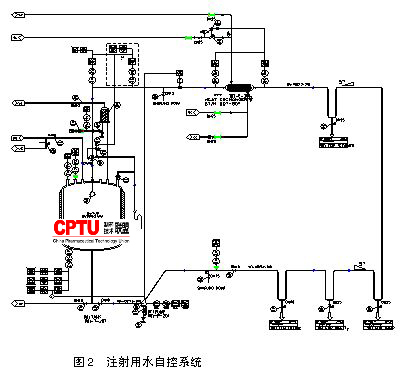
无菌原料药的无菌工艺验证探讨
瞬间微生物检测技术与应用
洁净区甲醛熏蒸效果验证
浅谈制药设备GMP验证
胶塞的热原测试方法
模板:YPJ-III型胶囊抛光机验证方案
]利用V模型验证ERP系统
药品生产管理之验证篇
多种剂型的认证要点
实验室纯水系统的认证要求
基于风险评估体系的厂房设计风险管理
基于风险评估体系的厂房设计风险管理
双人复核”的意义
OOE、OOT和OOS的解释
厂房、生产设施和设备多产品共用可行性风险评估报告
激素类药品与非激素类药品公共生产线生产激素类药品风险评估报告
最完美的ICHQ9诠释
风险评估技术在无菌粉针剂工程应用
图片附件: 7292825.jpg (2015-3-14 17:37, 24.87 KB) / 该附件被下载次数 13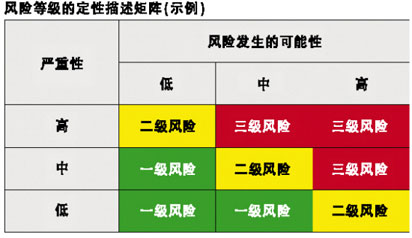 图片附件: 7292884.jpg (2015-3-14 17:37, 36.32 KB) / 该附件被下载次数 8
图片附件: 7292884.jpg (2015-3-14 17:37, 36.32 KB) / 该附件被下载次数 8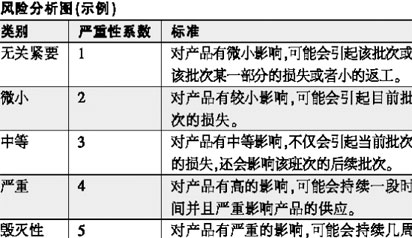
大输液的参数放行实践
参数放行讨论稿
质量源于设计
实施“质量源于设计”的五个关键因素
过程检测技术中的在线pH值测量
图片附件: 11144664.jpg (2015-3-14 17:53, 5.34 KB) / 该附件被下载次数 23 图片附件: 11144699.jpg (2015-3-14 17:54, 6.89 KB) / 该附件被下载次数 11
图片附件: 11144699.jpg (2015-3-14 17:54, 6.89 KB) / 该附件被下载次数 11 图片附件: 11144895.jpg (2015-3-14 17:54, 6.77 KB) / 该附件被下载次数 11
图片附件: 11144895.jpg (2015-3-14 17:54, 6.77 KB) / 该附件被下载次数 11
[size=3]设计质量高于一切
QbD是我国制药业追赶国际水平的重要机会
从一家跨国药企透视质量源于设计理念的实施
QbD被认为是国际商机通道 获FDA积极推动
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |