顺便说一下,如果是新手,刚开始接触引物设计,推荐使用Primer Premier 5.0,因为它界面简单,易学易用;如果你想把引物设计得尽善尽美,公认的首选软件是Oligo,其次我认为是DNAstar。Oligo功能强大,所以使用起来就没有Primer Premier 5.0那么简便。先用Primer Premier 5.0设计,然后把设计好的引物拿到Oligo里去检测这对引物的优劣,我想这对大多数引物设计者是一个不错的选择!
正在读primer premier 的 primer design guideline cuturl('http://www.premierbiosoft.com/tech_notes/PCR_Primer_Design.html')
结合你文中所提问些问题:
1。Guideline里说“Primers with melting temperatures above 65oC have a tendency for secondary annealing.” 这个“secondary annealing”如何理解?
2。GC Clamp和3端稳定性:可以这样简单理解吗:5端要GC多,3端要GC少。 Guideline里讲3端最后5个碱基里要避免3个G或3个C。但是对5端没有谈。
3。发卡结构:Guideline里说Optimally a 3' end hairpin with a ΔG of -2 kcal/mol and an internal hairpin with a ΔG of -3 kcal/mol is tolerated generally。其实第一还是G/C在3端的比例问题,第二是G/C配对问题。作者: 阿拉蕾 时间: 2011-8-12 10:49
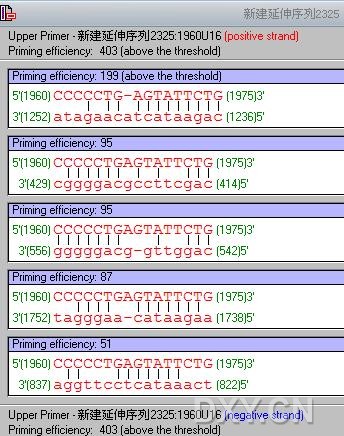
楼主你好,我用P5设计出来引物后,用Oligo 检测,最后得出的结果是Excessive difference between product and primer melting temperatures.
请问这个影响大吗?我可以用这对引物吗? 作者: @木木@ 时间: 2011-8-12 11:08
 图片附件: 1.jpg (2011-8-12 10:56, 67.77 KB) / 该附件被下载次数 26
图片附件: 1.jpg (2011-8-12 10:56, 67.77 KB) / 该附件被下载次数 26