标题:
【求助】融合PCR
[打印本页]
作者:
zhezhe
时间:
2015-7-2 21:26
标题:
【求助】融合PCR
我有两段基因,分别是1700bp和500bp左右,我希望将两段基因通过中间的重叠互补区域连接,再以此为模板进行PCR,这个是不是就叫融合PCR啊??哪里能找到这样的资料呢??我 扩了好久都没有目的条带,反而有一非目的条带很浓很浓,我真的好怕因为这个毕不了业。还挨老板骂。。。。。大家能帮帮我吗??我决得我的引物设计没有多大问题啊。不知道我应该如何检测我的引物合理与否呢??我现在几乎不知道是否该重设引物?不知道失败在哪里!帮帮我吧!谢谢大家。
作者:
nikonun
时间:
2015-7-2 21:27
挨老板骂算什么呀,毕不了业才是大事哦。
楼主你有更详细的资料吗?比如目的基因序列,引物序列,重叠序列,反应条件等等。
作者:
jiushikeshui371
时间:
2015-7-2 21:27
有,两段目的基因分别是
gaa gcatataaga gtgagattgc tcatcggtac
121 aatgatttgg gagaagaaca tttcagaggc ctggtgctgg ttgccttttc tcagtatctc
181 cagcagtgtc catttgagga tcatgtgaaa ctagccaagg aagtgactga gtttgcaaaa
241 gcctgtgctg ctgaagagtc aggggccaac tgtgacaaat cccttcacac cctgttcggg
301 gacaagctgt gcacggtggc ctccctccgg gacaagtacg gggacatggc cgactgctgc
361 gagaagcagg agcccgacag gaacgagtgc ttcctggcgc acaaggacga caacccgggc
421 ttccccccgc tggtggcccc cgagcccgac gcgctgtgcg ccgccttcca ggacaacgag
481 cagctgttcc tggggaagta cctgtacgaa attgccagaa gacatccgta cttctacgcc
541 ccagaactcc tgtactatgc tcaacagtat aaaggagtct ttgcggagtg ctgccaggcc
601 gcagacaagg ccgcctgcct gggacccaag attgaggctt tgagggaaaa agtactgctt
661 tcatctgcca aggagagatt caagtgtgcc agcctccaaa aattcggaga tagagccttt
721 aaagcctggt cagtagctcg cctgagccag cgatttccca aagctgactt tgcagagatc
781 tccaaggtgg tgacagatct taccaaagtc cacaaggaat gctgccatgg tgacctgctg
841 gagtgtgcag atgacagggc ggatcttgcc aagtatatgt gtgaaaatca agattcaatc
901 tccactaaac tgaaggaatg ctgtgataag cctgtgttgg aaaaatccca gtgtcttgct
961 gaggtggaaa gagatgagtt acctggtgac ctgccctcat tagctgctga ttttgttgaa
1021 gataaggagg tttgcaaaaa ctatcaggag gcaaaggatg tgttcctggg cacgtttttg
1081 tatgaatacg caagaaggca tccagagtac tctgtctcat tgcttttgag actcgccaag
1141 gaatatgaag ccacactaga gaaatgctgt gccaccgatg atcctcctac atgctatgcc
1201 aaagtgcttg atgaatttaa acctcttgtg gatgagcctc agaatttagt caaaacaaac
1261 tgtgaacttt ttgaaaaact tggagagtat ggcttccaaa atgcgctctt agttcgttac
1321 accaagaaag caccccaagt gtcaactcca actctcgtgg aggtctcaag aaaactagga
1381 aaagtgggca ccaaatgttg taagaaacct gaatcagaga gaatgtcctg tgctgaagac
1441 tttctgtccg tggtcctgaa ccggttgtgt gtgttgcacg agaagacccc agtgagcgag
1501 agagttacca aatgctgctc agagtccttg gtgaacagac gaccatgctt ttctggtctg
1561 gaagtcgatg agacctatgt tcccaaagag tttaatgctg aaacattcac tttccatgca
1621 gatttatgca cacttcctga ggctgagaaa caagtcaaga aacaaactgc acttgttgaa
1681 ctgctgaaac acaagcccaa ggcaacagat gaacaactga aaactgttat gggagatttt
1741 ggagcctttg tagagaagtg ctgcgcagct gaaaataaag agggctgctt ttctgaagag
1801 ggtccaaaac tcgttgctgc tgctcaagct gccttagtc
第二段:
TgtgatTTGcctCAAactcatGGTTTGTTGaatAGAAGAgccttgACTTTGTTGGGTcaaatgAGAagaTTGcctgccTCTtccTGTCAAaaggacagaaatgacttcgccttcCCACAAgacGTTttcggtGGTgacCAAtcccacaaggcccaagccTTGTCTGTTGTTcacGTTACTaacCAAaagatcttccacttcttcTGTacaGAGGCTtccTCTtctgctgcttggaacaccaccTTGTTGGAGgaGttcTGTACTGGTTTGgatAGACAATTGaccAGATTGgaGgcctgtgtcGTTCAAGAGGTTGGTGAGGGTGAGgctCCATTAACAaacGAGgactccatcTTAAGAaactacttccaaagaTTAtccTTAtacTTAcaaGAGaagaaatacTCTccttgtgcctggGAGatcgtcagagcagaGatcatgagatccttgtattattcatcaacagccttgCAAaaaagattaAGATCTGAGaaatga
四条引物分别是:
Primer1: csa上游:
5’ GAGGAATTC gaa gcatataaga gtgAGA (28) 3’
Primer2: csa下游:
5’ TCCACCACCT GAACCTCCTG AACC gactaaggcagcttgag 3’( 41bp)3’
Primer 3: IFNa上游
5’GGTTCAGGAG GTTCAGGTGG TGGA tgtgatTTGcc 3’
Primer 4 : IFNa下游
5’ TTT CGCCGGCG TCATTTCTCAGATC(25bp) 3’
重叠序列就是二三条间的
GGTTCAGGAG GTTCAGGTGG TGGA
我真的希望有谁能帮帮我,我的QQ是150503898,希望能给我更快更多帮助.....
作者:
eve_49
时间:
2015-7-2 21:27
这类试验我做的很少,以我看来,引物 1 可以。
引物 4 的 5’ TTT CGCCGGCG 是什么呢?是 要利用 GCCGGC 作酶切吗?如果是这样,那么 引物 4 中与模板互补的序列长度就只有 14 个碱基,是不是太短。
引物 3 也有同样的嫌疑,互补序列太短,只有 11 个碱基。
引物 2 中,模板互补区也不够长。
在我的试验里,引物中与模板互补区 20-22 个碱基。
另外还有一个千万要注意的地方,第一轮 PCR 时一定要用 Pfu 酶来扩增,因为它不会在 产物 的 3’ 末端加 A , 否则会造成最终产物的移码突变。
作者:
zhezhe
时间:
2015-7-2 21:28
就是用 GCCGGC (NotI)作酶切,引物3和4中如果我增加引物与模板的互补序列,那么就会出现引物二级结构(自连),我该着重注意什么呢?另外,takara 公司的LAtaq酶会不会给片段加A啊??
作者:
987789
时间:
2015-7-2 21:28
如果互补序列太短,PCR 会有困难,应该长一点,首先要保证PCR出条带,(不知道引物 1,2 PCR情况如何?)自连引物可以在做切胶回收这一步的时候去除。
不知道 LAtaq 是否会加 A ,这里别的战友应该能有知道的。 不过 LA 很贵的哦,你老板舍得吗 ? ^_^
作者:
33号
时间:
2015-7-2 21:30
我最近刚好也做了这样的试验,严格说应该叫做重组PCR。前些天测序后已经证明两个片断已经通过18bp的街头连在一起。
根据我的经验,我建议楼主这样做:
1.关于四条引物的设计:
a. csa上游:楼主的引物应该没问题。
b. 下游csa:从5到3依次为:街头+CSA下游。关于街头长度,18bp已经够用了,楼主用了24个bp也没有问题;CSA下游引物楼主短了点,17个bp也可以,最好在20bp以上。
c. IFNa上游: 从5到3依次为:街头+IFNa上游。IFNa上游引物楼主短了点,最好在20bp以上。
d. IFNa下游: 没有问题
2.关于PCR, 我建议楼主这样做:
a. 先分别扩出CSA和IFN的目的片断。胶回收。25cycles
b. 重组PCR楼主可以有两种方法:
1.将回收的两个片断,分别取5-10ul,加上其他PCR组分至30ul,注意不加任何引物。PCR 10 cycles。然后取5-10ul上述PCR的产物,加PCR组分至30ul,注意加入csa上游和IFNa下游引物。30 cycles.
2. 也可以把第一种方法两个PCR反应一起做,即直接在第一个PCR反应中加入引物。30cycles。两个方法我都能扩出目的片断。
3.关于酶:我用的是KOD plus, toyobo公司的。楼主的这个酶会有A加入,建议换。
楼主要先把两个目的片断扩出来。如果楼主两个片断已经分别克隆到载体上,两个片断第二轮PCR时连不上,我建议楼主可以把csa上游引物和 IFN下游引物分别设计两个,一个用载体的序列,一个用目的片断的,这样先用载体引物扩出目的片断,回收,然后再用片断引物作重组PCR,有点类似套式PCR。
好运!
作者:
zhezhe
时间:
2015-7-2 21:31
我已经很久没有开心过了。整天PCR。没有进展总觉得是自己的罪过......后面的路还长,不知道还有什么荆棘,我觉得我摄取知识的手段很差,希望你们也能从某些方面给点经验.....
谢谢以上两位的帮助。我的两个片段是单独的。没有连载体。之前我将两个片段分别P出来后,取出两个片段各2ul,加入PCR组分到25ul,但不加引物,10个循环后加入引物再进行25个循环,但是仍然没有结果。也试过同时加引物,都不行。不知道怎么摸条件了... ...我的引物已经设过了,加长了,但还是没有连上,甚至分别P两段都不那么容易了。
作者:
mod=8048
时间:
2015-7-2 21:32
Dr.dingo 长于 overlap PCR Mutagensis
可以电话咨询
作者:
changlhsyo
时间:
2015-7-2 21:32
1.“之前我将两个片段分别P出来后,取出两个片段各2ul” 回收了吗?
2.“10个循环后加入引物再进行25个循环” ,该引物可以设计一对内引物看看,上游第15个碱基开始,下游也前推15个。
3.实在不行楼主可以先把基因克隆到载体,测序真确后在行融合。
作者:
zhezhe
时间:
2015-7-2 21:32
分别P出来的片段回收了的,我现在准备通过酶切位点的方法相连.那么我想再问问.可以三片段一起连吗??就是两个小片段.加载体.放一个体系里面连.
作者:
lgm
时间:
2015-7-2 21:32
可以的。最好先过夜连接,第二天早上再补点酶,然后下午转化。不要用平头的。载体要选短点的。记得做自连的对照。
作者:
zhezhe
时间:
2015-7-2 21:34
我的三个酶分别是:EcoRI和BamHI, BamHI和NotI,我的载体是pPIC9K,有9千多BP,您看能一起连吗?另外您能帮我看一下能不能分别双酶切呢?我用NEB的酶,但找不到共用Buffer ....我好害怕切不下来,连不上,因为我的模板快用完了,再PCR我怕就没有了.....问题好象很多呢
作者:
standbyme
时间:
2015-7-2 21:35
你是想做酵母表达吗?你用BamH1酶切,不是把信号肽也切了吗?
你还是重选一个载体吧,比如pUC、pBlu。你最好先把两个片断分别连到载体上,然后从载体上把片断切下来,再连到载体上。这样做更加容易些,因为直接用PCR产物酶切连载体效率不高,何况是三个连。我以前三个片断连就是这样连上的。
作者:
standbyme
时间:
2015-7-2 21:35
双酶切还是一起切的好。你选buffer不一定要求每个酶多是100活性,活性有20-50%也可以了。NEB酶不错。
作者:
dreaming
时间:
2015-7-2 21:36
线粒体007大侠你好,我最近也在做重组PCR,为了把1560bP和861bp的两个目的片段连接起来,我前面已经分别扩增出两个目的基因并已经胶回收,这两天在跑第二轮PCR,也就是用P1和P4为引物去跑,PCR结果有出现目的条带,但是特异性不是很强,反而是在1200处有个非目的条带很亮,这种情况下我该改变什么条件进行优化,以提高我目的产物的特异性呢?希望能给予指导,万分感谢!!!
作者:
chengjie79
时间:
2015-7-2 21:37
您好!我是NEB(北京)的技术支持。
EcoRI和BamHI, BamHI和NotI可以双酶切。
EcoRI和BamHI用EcoRI的buffer,BamHI和NotI用BamHI的buffer就可以了,关于双酶切,您可以用Double Digest Finder软件,网址为cuturl('http://www.neb.com/nebecomm/DoubleDigestCalculator.asp?')使用很方便。
连接可以用16度过夜,如果不放心,可以高浓度T4,您先试试
作者:
dotaaa
时间:
2015-7-2 21:37
你好,我想问一下,BamH1和Hind111,BamH1和Xbal1可以一起双酶切吗,还是要分步来,它们有无共同的缓冲液?谢谢
作者:
dotaaa
时间:
2015-7-2 21:38
我想问一下,BamHI和HindIII,BamHI和XbalI能不能一起双酶切?它们有无共同的缓冲液?还是要分步酶切,谢谢!
作者:
baidukk
时间:
2015-7-2 21:38
你能不能把你第二轮PCR说得详细一点。还有你重叠区的长度。最好附张图
作者:
zhenxin
时间:
2015-7-2 21:39
相关疾病:
先天性卵巢发育不全综合征
线粒体大侠,我的引物是这么设计的:A基因:
上游与开放阅读框起始密码子附近的序列相对应,并在5 ’末端添加酶切位点和3个保护碱基;下游与B基因终止密码子附近的序列及A基因5’末端起始密码子附近的序列互补,并去除A基因的终止密码子TAA
上游:5’ CCCAAGCTT ACCATGGATGTCGTCGAGCAGC 3’ 31
下游:5’ GTAGCCGACCGCCATAATGGAAGC GATGGTCTTCTGCTTCTTGGG 3’ 45
B基因:
上游与A基因终止密码子附近的序列及B基因5 ’端起始密码子附近的序列相对应,同样去除A基因终止密码子TAA;下游与B基因的开放阅读框终止密码子序列互补,并在5’末端添加酶切位点和2个保护碱基。
上游:5’ CCCAAGAAGCAGAAGACCATCGCTTCC ATTATGGCGGTCGGCTAC 3’ 45
下游 5’GCTCTAGA TCAGGCAGCGCGCTGC 3’ 24
每条引物空格前为酶切位点和保护碱基或用于融合的互补区,其中互补区最后六个为引入的接头,其余前面的为B头或A尾的互补序列(上面文字中的说明).第二论PCR我采用的条件为:94度4min,94度1min,55度1min,72度2min,5个循环,然后94度1min,65度1min,72度2min,28个循环,72度10min.
作者:
remonte
时间:
2015-7-2 21:39
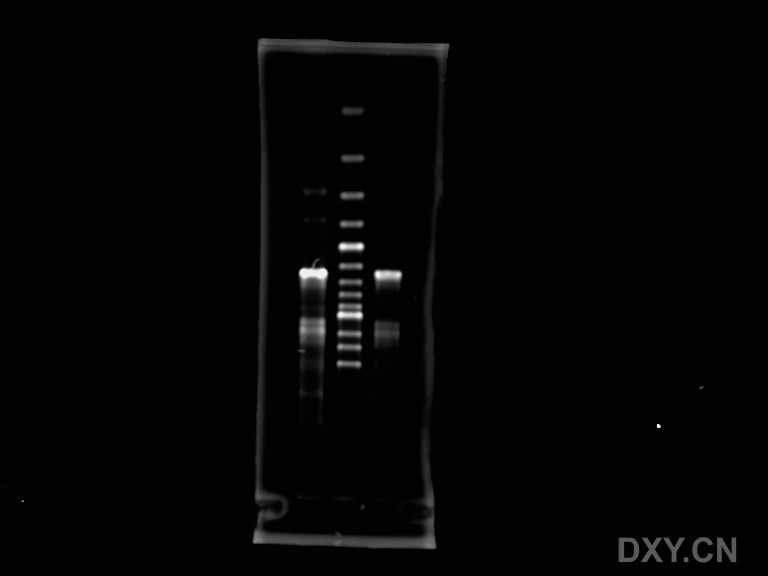
这是我第二轮PCR做出来的电泳图
图片附件:
47953622.jpg
(2015-7-2 21:39, 16.14 KB) / 该附件被下载次数 5
http://bbs.antpedia.com/attachment.php?aid=34381
作者:
remonte
时间:
2015-7-2 21:40
点样孔在下,跑得最慢的那条是我想要的,其上一条弱带,以及最前面那条很亮的都不是目的条带,请问我该如何优化条件而获得特异性强的目的条带?
作者:
JK.jon
时间:
2015-7-2 21:41
1."5个循环" 先提高到十个看看,然后取出1ul做模板加引物再跑一次PCR。
2. 把你要的那个条带切下来,然后设计一对引物再扩。该对引物要从你原先的引物位点后开始。因为你的目的条带比较模糊,不要用原来的引物,否则会扩出一片带出来。
3.优化退火温度,如果没有梯度PCR很麻烦。你不要把退火温度调这么高,先从55度试,然后1-2度梯加。
作者:
xevin
时间:
2015-7-2 21:42
您好,很感谢您提供的双酶切信息,我查过你们的网站,但我还就是有点担心 酶切效果,因为我后面要进行连接,总担心共用BUFFER会影响效果,不知道是否该提高酶切时间呢?
作者:
xevin
时间:
2015-7-2 21:42
大侠您好,很感谢您的帮助,我其实很不想做酵母,因为我们实验室做酵母都没有成功过,而且酵母很麻烦,我担心。但是我不知道我的蛋白应该在酵母里面还是在大肠里面表达,您看,我的基因总共有2300BP左右,在大肠里面是不是不大可能表达?另外,我选用的BamhI这个位点是为了将我的两个片段连上,因为我用PCR的方法始终融合不上,所以我就在LINKER后用酶切位点相连。连上后就不用那个位点了。你看......
作者:
xevin
时间:
2015-7-2 21:43
我现在有三个问题,自己解决不了:
1. 我不知道自己的蛋白在大肠还是酵母里面表达比较好,酵母好麻烦。
2. 有谁能告诉我血清白蛋白(我用的狗血清白蛋白)的结构,功能区域吗?我想取白蛋白的某一部分和我的基因融合,从而提高我的蛋白作用,但我不知道取哪一段,甚至不知道哪里去查资料....
3. 关于酵母表达,我用PPIC9K ,需要注意些什么吗?
我的问题有点初级,如果有什么没有写清楚,望大家提出,希望能得到大家的帮助.....
作者:
zhezhe
时间:
2015-7-2 21:43
很感谢您提供的双酶切信息,我查过你们的网站,但我还就是有点担心 酶切效果,因为我后面要进行连接,总担心共用BUFFER会影响效果,不知道是否该提高酶切时间呢?
作者:
zhezhe
时间:
2015-7-2 21:45
很感谢您的帮助,我其实很不想做酵母,因为我们实验室做酵母都没有成功过,而且酵母很麻烦,我担心。但是我不知道我的蛋白应该在酵母里面还是在大肠里面表达,您看,我的基因总共有2300BP左右,在大肠里面是不是不大可能表达?另外,我选用的BamhI这个位点是为了将我的两个片段连上,因为我用PCR的方法始终融合不上,所以我就在LINKER后用酶切位点相连。连上后就不用那个位点了。你看......
作者:
zhezhe
时间:
2015-7-2 21:45
我现在有三个问题,自己解决不了:
1. 我不知道自己的蛋白在大肠还是酵母里面表达比较好,酵母好麻烦。
2. 有谁能告诉我血清白蛋白(我用的狗血清白蛋白)的结构,功能区域吗?我想取白蛋白的某一部分和我的基因融合,从而提高我的蛋白作用,但我不知道取哪一段,甚至不知道哪里去查资料....
3. 关于酵母表达,我用PPIC9K ,需要注意些什么吗?
我的问题有点初级,如果有什么没有写清楚,望大家提出,希望能得到大家的帮助.....
作者:
dmg
时间:
2015-7-2 21:46
Q1:按照我的经验,你这么长的片段很难在酵母里表达,何况你的序列也没有药酵母密码子优化。建议你还是在原核表达,应该可以表达的。
Q3:酵母表达操作也不是很难,用PPIC9K是可以用G418筛选高拷贝的重组子,通常情况下,有1-10%的高拷贝重组子,如果你用2mg/ml的G418筛选可以从数万个重组子中筛选出几十个高拷贝的。
Q2:无法回答。
另外:建议还是找一个短点的载体。
作者:
zhezhe
时间:
2015-7-2 21:47
我就是因为片段太长,所以害怕在原核里面表达不了.另外,我想干脆两种一起做,你看呢... ...
1. 但是我不知道如果我在原核里面表达,我用什么载体好点呢?
2. 我没有G418,而且担心那种抗生素有点贵,怕老板不愿意为我一个人买,所以您看能不能在没有G418的情况下筛选呢?
作者:
zhezhe
时间:
2015-7-2 21:47
还有,我的后面段基因是优化过了的.
作者:
dmg
时间:
2015-7-2 21:47
这样长的片段在原核表达没有问题的,pET、pGEX系列的载体都是比较常用的原核表达载体,这两种载体都是融合表达,在目的蛋白上会多出一些氨基酸序列,但是比较好表达,不知道你表达后是想做什么用?
我想只有一段优化不会提高表达效果的。G418贵了点,但是现在好像便宜了。不筛高拷贝的,你只能多挑克隆,“海选”了。
作者:
zhezhe
时间:
2015-7-2 21:48
我做融合表达的目的是为了与单独表达时进行一些蛋白功能比较.
海选的意思是我需要挑很多克隆进行表达,看哪个的表达量更高吗?我们实
验室做酵母的都失败了,不知道我的结果会怎样......
欢迎光临 分析测试百科 (http://bbs.antpedia.com/)
Powered by Discuz! 5.5.0
 图片附件: 47953622.jpg (2015-7-2 21:39, 16.14 KB) / 该附件被下载次数 5
图片附件: 47953622.jpg (2015-7-2 21:39, 16.14 KB) / 该附件被下载次数 5