当前,SNPs研究在国际上有很大的进展,NATURE、Science、New England 、PNAS等顶尖杂志在近几年发表了大量的论文,国内各种医学权威期刊几乎每期都刊登关于SNPs的论文。当然,国内与国外相比还是有很大差距的,这体现在国内的小样本、研究内容不深入(仅限于关联研究)、基因分型技术手段落后等等方面。众所周知,研究生是科研的主力军,大多数论文工作都是由我们研究生完成,因此,本版块特开辟此专帖,大家集中讨论关于SNPs研究的一些最新进展,希望大家积极参与,互相促进,在此热贴下跟帖者会给予优先奖励。
此次讨论共分5个方面,大家可以选择一个侧面或者多个侧面阐述之,主要包括:
1. SNPs的基础知识介绍(SNP basics, definition)
2. SNPs的分型方法(SNP genotyping methods)
3. 如何选择研究的SNPs(Selection of targeted SNPs)
4. 如何采用分子流行学的方法对SNPs进行研究 (Association studies using molecular epidemiology)
5. SNPs的功能学验证(Identification of Functional SNPs)
(single nucleotide polymorphism , SNP,发音为“snips”), 主要是指在基因组水平上由单个核苷酸的变异所引起的 DNA 序列多态性。它是人类可遗传的变异中最常见的一种。占所有已知多态性的 90% 以上。 SNP 在人类基因组中广泛存在,平均每 500 ~ 1000 个碱基对中就有 1 个,估计其总数可达 300 万个甚至更多。
SNP 所表现的多态性只涉及到单个碱基的变异,这种变异可由单个碱基的转换 (transition) 或颠换 (transversion) 所引起,也可由碱基的插入或缺失所致。但通常所说的 SNP 并不包括后两种情况。
理论上讲, SNP 既可能是二等位多态性,也可能是 3 个或 4 个等位多态性,但实际上,后两者非常少见,几乎可以忽略。因此,通常所说的 SNP 都是二等位多态性的。这种变异可能是转换 (C T ,在其互补链上则为 G A) ,也可能是颠换 (C A , G T , C G , A T) 。转换的发生率总是明显高于其它几种变异,具有转换型变异的 SNP 约占 2/3 ,其它几种变异的发生几率相似。 Wang 等的研究也证明了这一点。转换的几率之所以高,可能是因为 CpG 二核苷酸上的胞嘧啶残基是人类基因组中最易发生突变的位点,其中大多数是甲基化的,可自发地脱去氨基而形成胸腺嘧啶。
在基因组 DNA 中,任何碱基均有可能发生变异,因此 SNP 既有可能在基因序列内,也有可能在基因以外的非编码序列上。总的来说,位于编码区内的 SNP(coding SNP,cSNP) 比较少,因为在外显子内,其变异率仅及周围序列的 1/5 。但它在遗传性疾病研究中却具有重要意义,因此 cSNP 的研究更受关注。
先形成的 SNP 在人群中常有更高的频率,后形成的 SNP 所占的比率较低。各地各民族人群中特定 SNP 并非一定都存在,其所占比率也不尽相同,但大约有 85% 应是共通的。
SNP 自身的特性决定了它更适合于对复杂性状与疾病的遗传解剖以及基于群体的基因识别等方面的研究:
SNP 数量多,分布广泛。据估计,人类基因组中每 1000 个核苷酸就有一个 SNP ,人类 30 亿碱基中共有 300 万以上的 SNPs 。 SNP 遍布于整个人类基因组中,根据 SNP 在基因中的位置,可分为基因编码区 SNPs ( Coding-region SNPs , cSNPs )、基因周边 SNPs ( Perigenic SNPs , pSNPs )以及基因间 SNPs ( Intergenic SNPs , iSNPs )等三类。
SNP 适于快速、规模化筛查。组成 DNA 的碱基虽然有 4 种,但 SNP 一般只有两种碱基组成,所以它是一种二态的标记,即二等位基因( biallelic )。 由于 SNP 的二态性,非此即彼,在基因组筛选中 SNPs 往往只需 +/- 的分析而不用分析片段的长度,这就利于发展自动化技术筛选或检测 SNPs 。
SNP 等位基因频率的容易估计。采用混和样本估算等位基因的频率是种高效快速的策略。该策略的原理是:首先选择参考样本制作标准曲线,然后将待测的混和样本与标准曲线进行比较,根据所得信号的比例确定混和样本中各种等位基因的频率。 易于基因分型。 SNPs 的二态性,也有利于对其进行基因分型。对 SNP 进行基因分型包括三方面的内容: (1) 鉴别基因型所采用的化学反应,常用的技术手段包括: DNA 分子杂交、引物延伸、等位基因特异的寡核苷酸连接反应、侧翼探针切割反应以及基于这些方法的变通技术; (2) 完成这些化学反应所采用的模式,包括液相反应、固相支持物上进行的反应以及二者皆有的反应。 (3) 化学反应结束后,需要应用生物技术系统检测反应结果。
SN P s 研究现在正处于发展之中, 虽然它有很好的前景, 但目前仍存在不少问题.
1.复杂疾病相关分析中的问题
在Skok lo ster 举行的国际SN P 及复杂基因分
析会议上透露出许多问题[ 2 ] , 利用SN P 寻找致病基因的工作并不象最初想象的那样简单。研究者除需要大量的SN P s, 有时需要弄清被研究人群的历史,如他们的迁移模式等, 在对心脏病危险因素L PL 基因的研究中, 由于减数分裂中的基因重组, 使SN P的关联分析很困难; Ro salind Harding 用SN P s 研究镰刀细胞贫血的B球蛋白基因时也遇到了困难,她认为研究者除依靠SN P 外,还需知道疾病的模式及被研究人群的历史。
2.技术问题
目前虽然有大量检测SN P 的方法, 但大都价格昂贵, 速度较慢, 这限制了其在法庭科学中的应用,所以迫切需要有低成本, 高生产率的新方法涌现。
3.SNP 命名问题
目前没有一个统一标准的命名方法, 这对于Y2SN P 来说尤为紧迫, 由于不同SN P 由不同实验室测定, 现在至少存在6 种不同的命名系统。作者: 快乐的大脚 时间: 2011-8-15 16:59
全基因组关联研究确实是最近的热点,高分的杂志发了很多这类文章。目前国际上对复杂疾病(complex disease)的态度现在是做关联了。但是太费钱了。也确实是一个框架性的工作。Nature. 2007 Jun 7;447(7145):661-78.
Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls.Wellcome Trust Case Control Consortium.作者: 快乐的大脚 时间: 2011-8-15 17:15
The Cancer Genome Atlas (TCGA) is a comprehensive and coordinated effort to accelerate our understanding of the molecular basis of cancer through the application of genome analysis technologies, including large-scale genome sequencing.
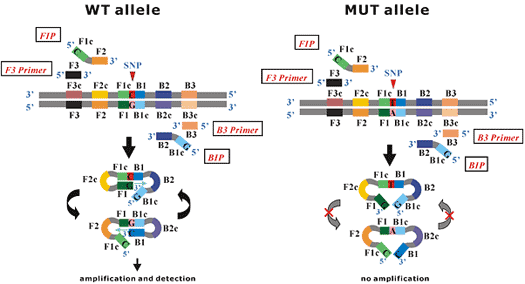
The figures below show the basic principles of the process when using Wild Type (WT) Primers.
The FIP and BIP are designed to contain a SNP nucleotide (in this case of Wild Type allele) at 5' end, respectively.
Using the WT primers, when the target gene is the WT allele, DNA synthesis from dumbbell-like starting structure proceeds and the LAMP amplification cycling continues. In contrast, when the target gene is the Mutant (MUT) allele, no DNA synthesis proceeds from dumbbell-like structure and the LAMP amplification cycling dose not occur. Even if DNA synthesis proceeds in one step due to miscopy, the amplification reaction is either halted in other steps or is delayed since repetition of this reaction continually checks at each cycling step of the DNA replication.
剩下的就是传统方法如酶切,pcr-sscp等,也有杂志接受,但一般需要测序验证
缺点工作量太大,结果不准确,不是所有位点都可以做,但成本很低
突变数据库
Human Gene Mutation Database (HGMD)
cuturl('http://www.uwcm.ac.uk/uwcm/mg/hgmd0.html')
The Genome Database (GD
cuturl('http://www.gdb.org')
SNP数据库
Database of Single Nuleotide Polymorphisms (dbSNP)
cuturl('http://www.ncbi.nlm.nih.gov/SNP/')
Human Genome Variation Database (HGVbase)
cuturl('http://hgvbase.cgb.ki.se/')
The Snp Consortium, LTD.(TSC)
cuturl('http://snp.cshl.org/')
Transferability of tag SNPs in genetic association studies
in multiple populations
这是发在nature gentics 上一篇很有意思的letter
证实了HapMap
DNA samples can be used to select tags for genome-wide
association studies in many samples around the world.
SNP genotype 和tag SNP的 方法里面也有介绍.作者: 快乐的大脚 时间: 2011-8-16 10:31
另外我有一个问题想向各位请教:我的课题是用PCR-SSP的方法分析4个位点的基因多态性,本来我4个位点的反应条件和反应体系均很稳定且高效,并且已经很顺利的完成了120份DNA标本4个位点的多态性分析,但是自此后,我其中一个位点就跑不出来,我起初怀疑引物出了问题(刚新稀释了引物,前面的用完了),于是先后从新合成了3遍(2次invitrogen,1次生工),并且彻底更换了所有试剂,并同时采用阴性和阳性对照,但结果仍无改观.我也怀疑过是DNA质量问题,但是同一批DNA跑其他3个位点都没问题.我几乎想尽了所有的办法,也向所里所有老师请教过,但都没有肯定的解释.昨天,我把以前该位点跑的好的DNA和跑不出来的DNA各5份加上阴性对照共11份做了比较,电泳结果显示:以前跑的好的DNA仍跑的很好,跑的不好的DNA仍没有跑出来,阴性对照显示没有污染.从这个结果看来很可能是DNA质量问题,但是其他3个位点却没有问题!这是什么原因呢?不知哪位师兄师姐有好的建议,请多指教!万分感激!
我用的DNA提取试剂盒是Qiagen GENTRA Puregene blood core kit B , Lot NO :GS22215.出问题前用的盒子是GENETRA Puregene blood kit , Lot NO : D-5000.厂家称GS22215与D-5000的组成完全相同,前者是后者换包装后的产品(品牌兼并的原因). 作者: 快乐的大脚 时间: 2011-8-16 10:35
最后,用我最近看到一篇文章中的一句话作为结束吧“The contribution of these rare variants to NHL susceptibility will likely only be assessed fully when higher throughput sequencing methods allow comprehensive sequencing of the whole gene in hundreds, or even thousands of NHL cases and controls.”
 图片附件: 63424848.gif (2011-8-15 17:22, 18.15 KB) / 该附件被下载次数 7
图片附件: 63424848.gif (2011-8-15 17:22, 18.15 KB) / 该附件被下载次数 7