 图片附件: 48368521_snap.jpg (2011-9-6 17:35, 14.7 KB) / 该附件被下载次数 15
图片附件: 48368521_snap.jpg (2011-9-6 17:35, 14.7 KB) / 该附件被下载次数 15 图片附件: 53545640_snap.jpg (2011-9-7 16:28, 62.05 KB) / 该附件被下载次数 14
图片附件: 53545640_snap.jpg (2011-9-7 16:28, 62.05 KB) / 该附件被下载次数 14 图片附件: 56874559_snap.jpg (2011-9-7 17:10, 62.05 KB) / 该附件被下载次数 12
图片附件: 56874559_snap.jpg (2011-9-7 17:10, 62.05 KB) / 该附件被下载次数 12 图片附件: 1.gif (2011-9-10 13:20, 19.51 KB) / 该附件被下载次数 9
图片附件: 1.gif (2011-9-10 13:20, 19.51 KB) / 该附件被下载次数 9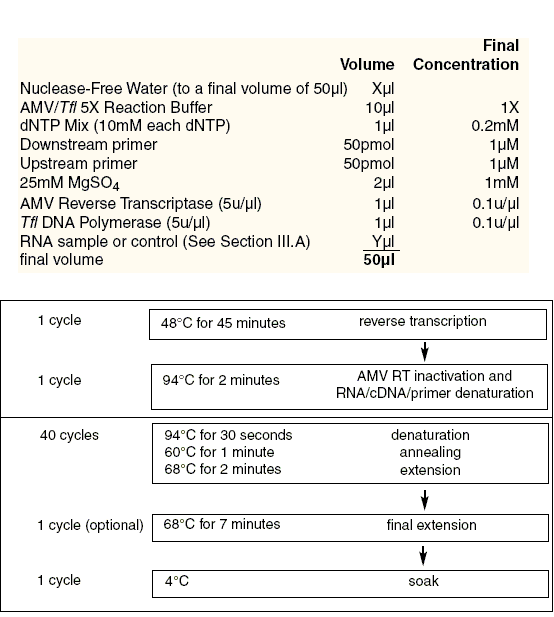 图片附件: 2.gif (2011-9-10 13:20, 19.51 KB) / 该附件被下载次数 11
图片附件: 2.gif (2011-9-10 13:20, 19.51 KB) / 该附件被下载次数 11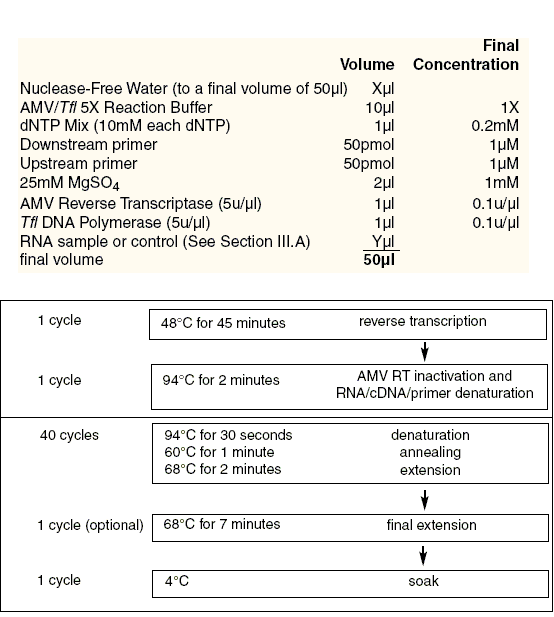 图片附件: 47712396.jpg (2011-9-10 13:32, 12.54 KB) / 该附件被下载次数 7
图片附件: 47712396.jpg (2011-9-10 13:32, 12.54 KB) / 该附件被下载次数 7 图片附件: 88545052.jpg (2011-9-10 13:49, 12.68 KB) / 该附件被下载次数 4
图片附件: 88545052.jpg (2011-9-10 13:49, 12.68 KB) / 该附件被下载次数 4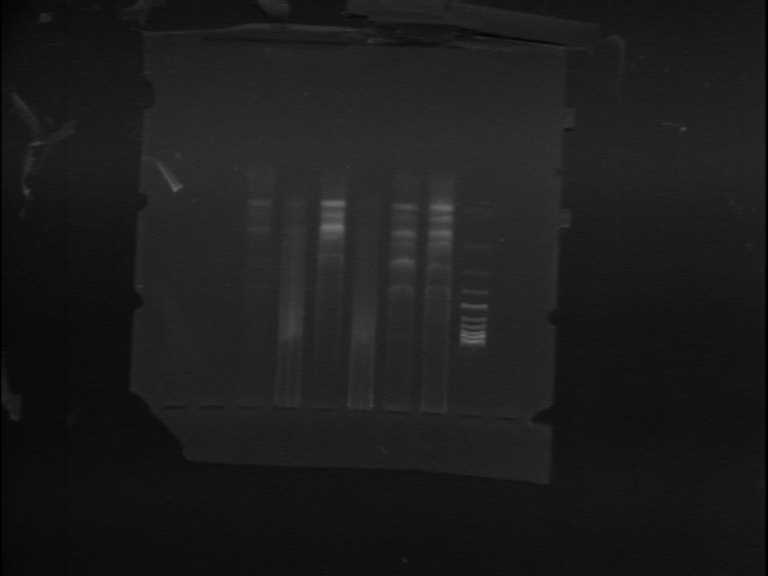 图片附件: 98596696.jpg (2011-9-13 18:00, 35.65 KB) / 该附件被下载次数 13
图片附件: 98596696.jpg (2011-9-13 18:00, 35.65 KB) / 该附件被下载次数 13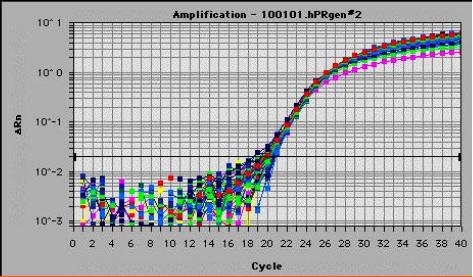 图片附件: 87932952.jpg (2011-9-14 15:10, 14.48 KB) / 该附件被下载次数 17
图片附件: 87932952.jpg (2011-9-14 15:10, 14.48 KB) / 该附件被下载次数 17 图片附件: 54637279.jpg (2011-9-14 16:34, 8.91 KB) / 该附件被下载次数 8
图片附件: 54637279.jpg (2011-9-14 16:34, 8.91 KB) / 该附件被下载次数 8 图片附件: 90407926.jpg (2011-9-14 17:55, 29.54 KB) / 该附件被下载次数 12
图片附件: 90407926.jpg (2011-9-14 17:55, 29.54 KB) / 该附件被下载次数 12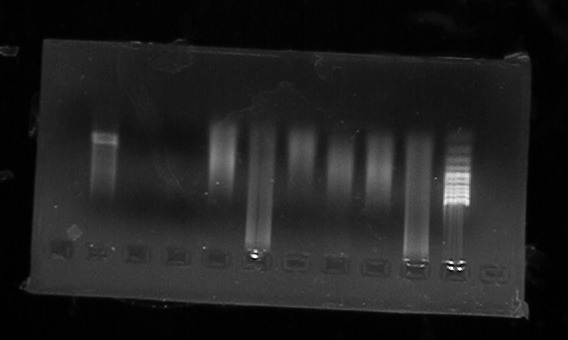 图片附件: 85029882.png (2011-9-15 11:30, 14.83 KB) / 该附件被下载次数 10
图片附件: 85029882.png (2011-9-15 11:30, 14.83 KB) / 该附件被下载次数 10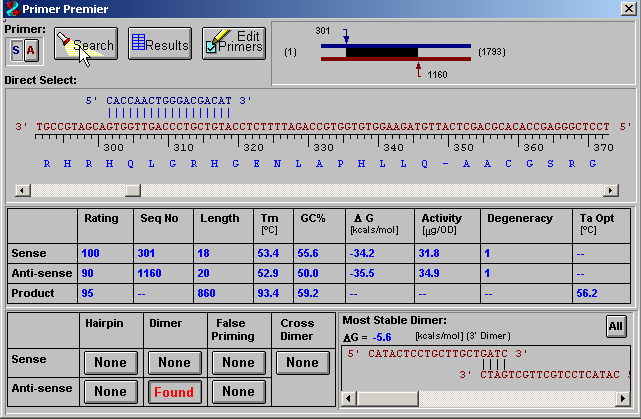 图片附件: 14830774.png (2011-9-15 11:31, 37.47 KB) / 该附件被下载次数 10
图片附件: 14830774.png (2011-9-15 11:31, 37.47 KB) / 该附件被下载次数 10
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |