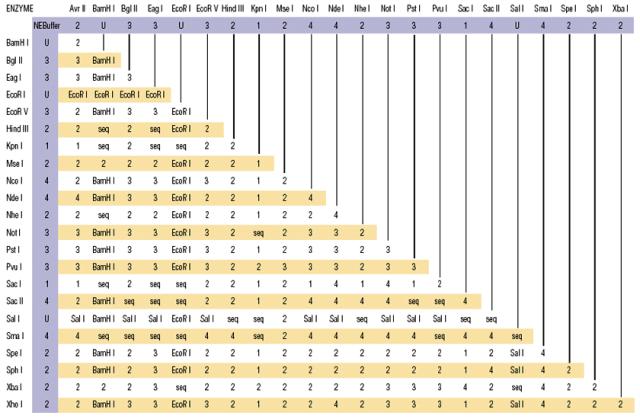
新英格兰公司提供的:双酶切buffer的选择
注:
U
Supplied with its own unique reaction buffer that is different from the four standard NEBuffers. Its compatibility with the four standard NEBuffers is indicated by the chart.
BSA
Supplied with a separate vial of bovine serum albumin (10 mg/ml). To obtain 100% activity BSA should be added to the 1X reaction mix to a final concentration of 100 µg/ml.
SAM
Supplied with a separate vial of S-adenosyl-methionine (SAM). To obtain 100% activity, SAM should be added to the 1X reaction mix as specified on the product data card.
dd
When performing a double digest with this enzyme, this NEBuffer is recommended because it minimizes star activity.
*
Purified by scientists at SibEnzyme and supplied with a SibEnzyme buffer (B, K, O, W or Y) which ensures 100% activity. Its compatibility with the NEBuffer System is indicated on the chart
NR
This buffer is not recommended for use with with this enzyme
Buffer Compositions
(1X): NEBuffer 1 (yellow): 10 mM Bis Tris Propane-HCl, 10 mM MgCl2, 1 mM DTT (pH 7.0 at 25°C). NEBuffer 2 (blue): 10 mM Tris-HCl, 10 mM MgCl2, 50 mM NaCl, 1 mM DTT (pH 7.9 at 25°C). NEBuffer 3 (red): 50 mM Tris-HCl, 10 mM MgCl2, 100 mM NaCl, 1 mM DTT (pH 7.9 at 25°C). NEBuffer 4 (green): 20 mM Tris-acetate, 10 mM magnesium acetate, 50 mM potassium acetate, 1 mM DTT (pH 7.9 at 25°C).
DetaiLed information :cuturl('http://circuit.neb.com/neb/tech/tech_resource/restriction/buffers/buffer_legend.html')
我想你的猜测有道理,Taq plus的保真效率高,适合长片段(10-30kb)扩增,公司的说明书上有这样的描述:
(1) high fidelity with an error frequency 1.6 / 106 (or 0.0016/103) during DNA synthesis.
(2) Taq Plus increases the efficiency of polymerization reaction, resulting in a great percentage of extenuation reaction complction up to 10 kb to 30 kb. Pfu has a temperature optimum between 72-78 ℃ and remains > 95% active following 1-hour incubation at 95 ℃.
 图片附件: 24515305_snap.jpg (2011-10-7 16:16, 48 KB) / 该附件被下载次数 4
图片附件: 24515305_snap.jpg (2011-10-7 16:16, 48 KB) / 该附件被下载次数 4