 图片附件: 72442305.jpg (2011-10-13 12:11, 48.52 KB) / 该附件被下载次数 16
图片附件: 72442305.jpg (2011-10-13 12:11, 48.52 KB) / 该附件被下载次数 16 图片附件: 43004303.jpg (2011-10-13 12:27, 53.68 KB) / 该附件被下载次数 14
图片附件: 43004303.jpg (2011-10-13 12:27, 53.68 KB) / 该附件被下载次数 14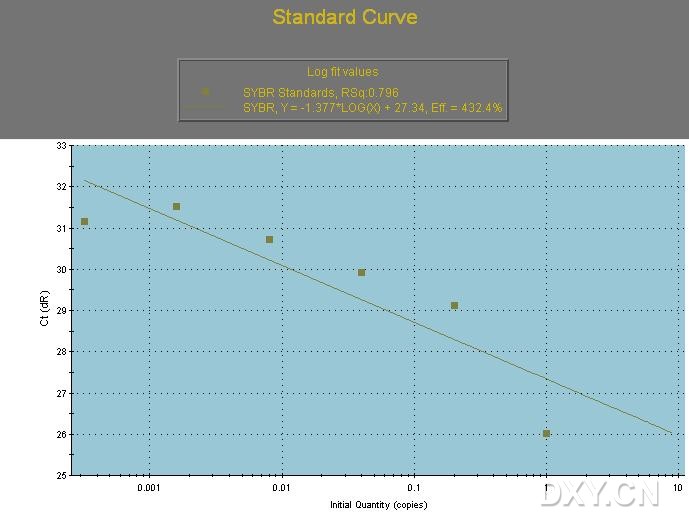 图片附件: 55813776_snap.jpg (2011-10-13 15:37, 69.84 KB) / 该附件被下载次数 5
图片附件: 55813776_snap.jpg (2011-10-13 15:37, 69.84 KB) / 该附件被下载次数 5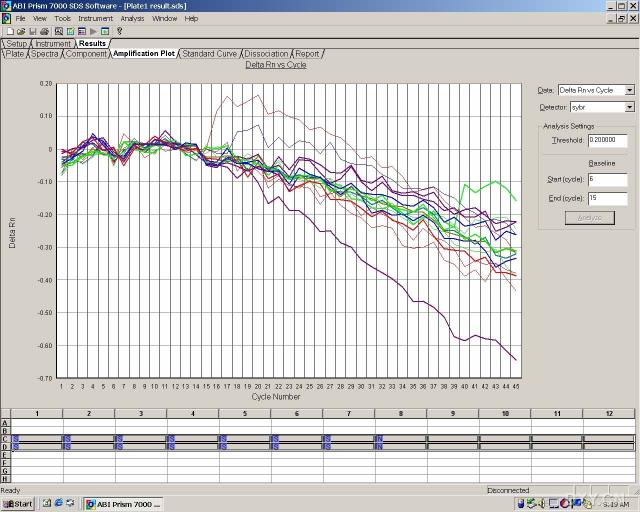 图片附件: 52530402_snap.jpg (2011-10-13 15:40, 56.56 KB) / 该附件被下载次数 4
图片附件: 52530402_snap.jpg (2011-10-13 15:40, 56.56 KB) / 该附件被下载次数 4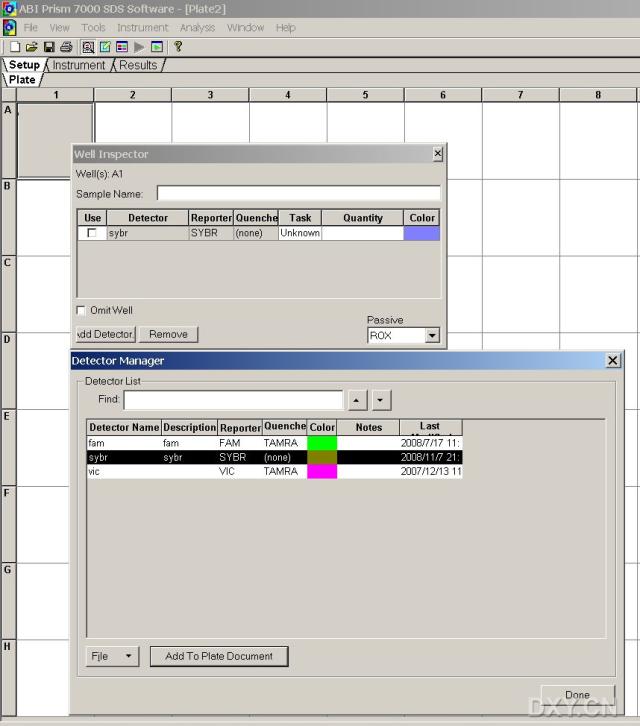 图片附件: 90261275_snap.jpg (2011-10-13 16:46, 69.94 KB) / 该附件被下载次数 11
图片附件: 90261275_snap.jpg (2011-10-13 16:46, 69.94 KB) / 该附件被下载次数 11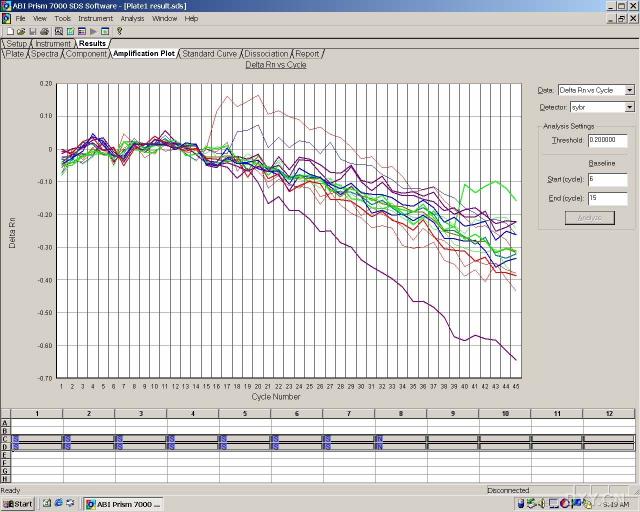 图片附件: 55292155.jpg (2011-10-13 17:00, 52.27 KB) / 该附件被下载次数 18
图片附件: 55292155.jpg (2011-10-13 17:00, 52.27 KB) / 该附件被下载次数 18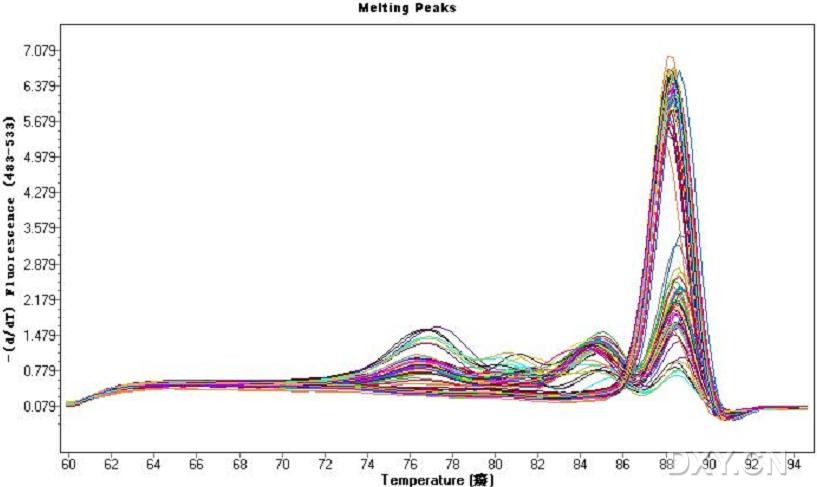 图片附件: 92322790.png (2011-10-13 17:29, 38.95 KB) / 该附件被下载次数 9
图片附件: 92322790.png (2011-10-13 17:29, 38.95 KB) / 该附件被下载次数 9 图片附件: 62311191.png (2011-10-13 17:34, 38.95 KB) / 该附件被下载次数 10
图片附件: 62311191.png (2011-10-13 17:34, 38.95 KB) / 该附件被下载次数 10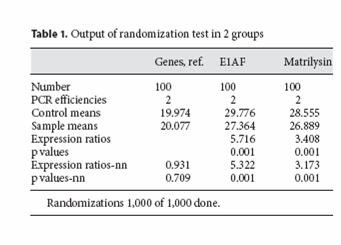 图片附件: 43299692.jpg (2011-10-14 15:58, 49.66 KB) / 该附件被下载次数 8
图片附件: 43299692.jpg (2011-10-14 15:58, 49.66 KB) / 该附件被下载次数 8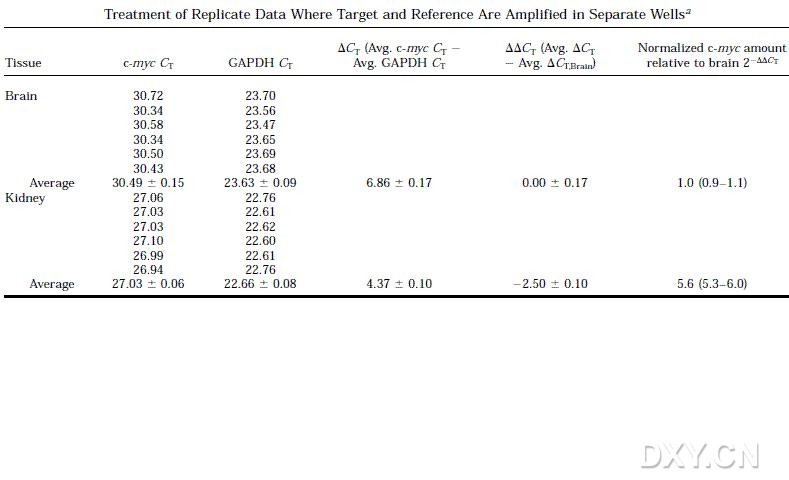 图片附件: 75930535.jpg (2011-10-14 16:50, 69.33 KB) / 该附件被下载次数 5
图片附件: 75930535.jpg (2011-10-14 16:50, 69.33 KB) / 该附件被下载次数 5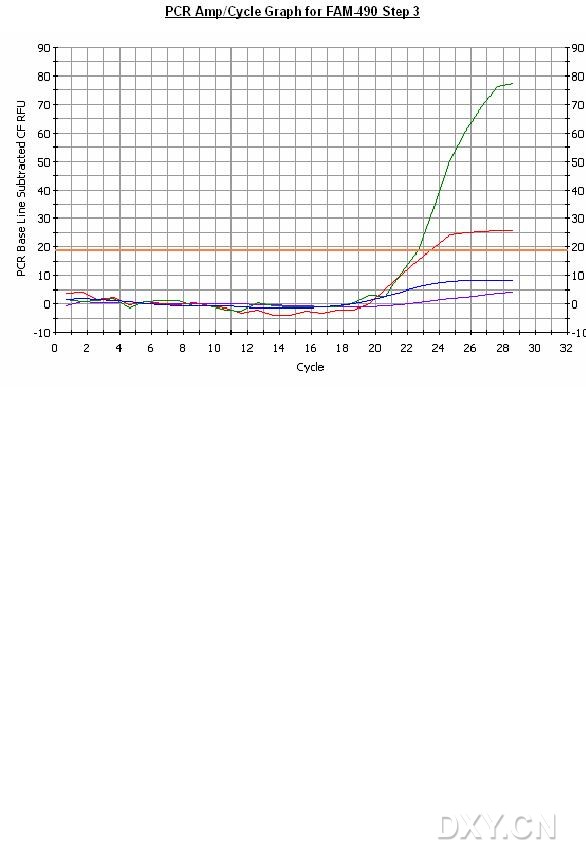 图片附件: 41321852.png (2011-10-14 16:57, 52.23 KB) / 该附件被下载次数 1
图片附件: 41321852.png (2011-10-14 16:57, 52.23 KB) / 该附件被下载次数 1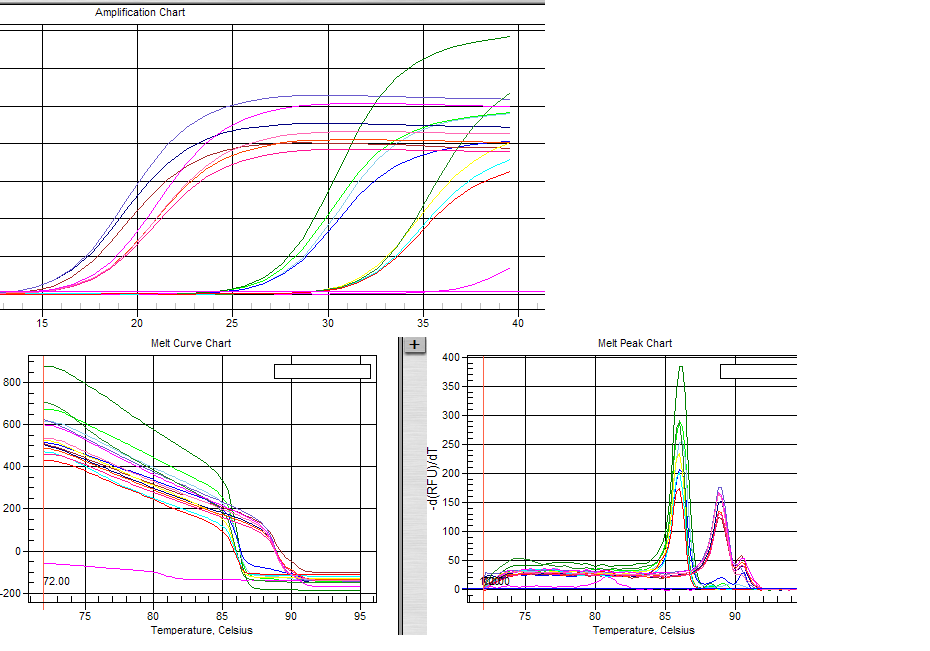 图片附件: 76955314_snap.jpg (2011-10-14 17:16, 37.15 KB) / 该附件被下载次数 2
图片附件: 76955314_snap.jpg (2011-10-14 17:16, 37.15 KB) / 该附件被下载次数 2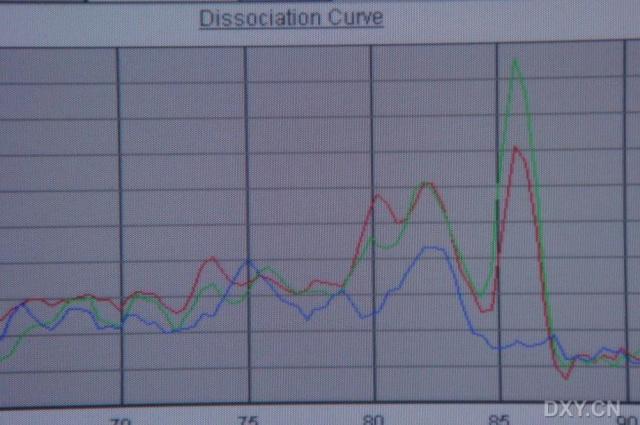 图片附件: 97332818.jpg (2011-10-15 09:21, 76.47 KB) / 该附件被下载次数 6
图片附件: 97332818.jpg (2011-10-15 09:21, 76.47 KB) / 该附件被下载次数 6 图片附件: 16116073_snap.jpg (2011-10-15 09:26, 23.94 KB) / 该附件被下载次数 10
图片附件: 16116073_snap.jpg (2011-10-15 09:26, 23.94 KB) / 该附件被下载次数 10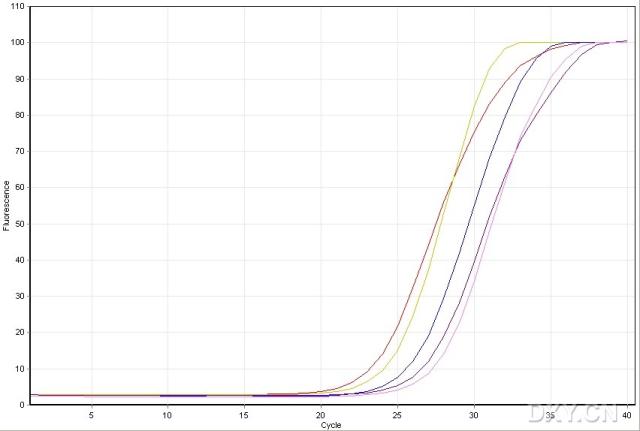 图片附件: 32954712.jpg (2011-10-15 09:26, 22.89 KB) / 该附件被下载次数 5
图片附件: 32954712.jpg (2011-10-15 09:26, 22.89 KB) / 该附件被下载次数 5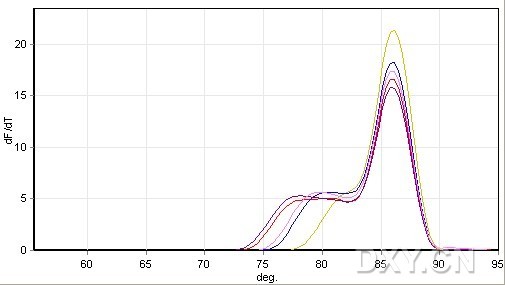 图片附件: 62809608.jpg (2011-10-15 09:27, 26.86 KB) / 该附件被下载次数 10
图片附件: 62809608.jpg (2011-10-15 09:27, 26.86 KB) / 该附件被下载次数 10 图片附件: 30279866.jpg (2011-10-15 10:37, 37.92 KB) / 该附件被下载次数 5
图片附件: 30279866.jpg (2011-10-15 10:37, 37.92 KB) / 该附件被下载次数 5 图片附件: 91450625_snap.jpg (2011-10-15 10:42, 81.49 KB) / 该附件被下载次数 3
图片附件: 91450625_snap.jpg (2011-10-15 10:42, 81.49 KB) / 该附件被下载次数 3 图片附件: 87436868.jpg (2011-10-15 11:36, 13.14 KB) / 该附件被下载次数 1
图片附件: 87436868.jpg (2011-10-15 11:36, 13.14 KB) / 该附件被下载次数 1 图片附件: 77902851_snap.jpg (2011-10-15 12:01, 25.77 KB) / 该附件被下载次数 2
图片附件: 77902851_snap.jpg (2011-10-15 12:01, 25.77 KB) / 该附件被下载次数 2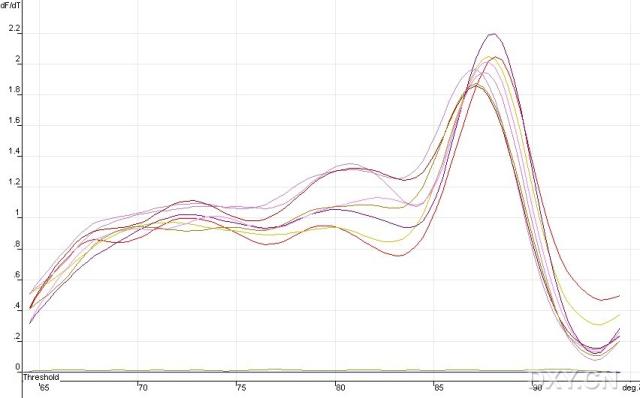 图片附件: 35255276.jpg (2011-10-15 12:02, 53.74 KB) / 该附件被下载次数 6
图片附件: 35255276.jpg (2011-10-15 12:02, 53.74 KB) / 该附件被下载次数 6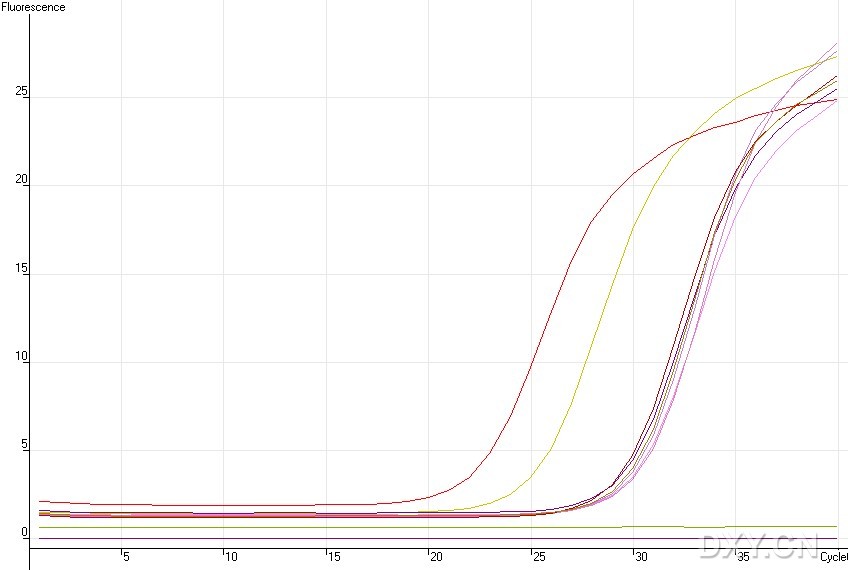 图片附件: 29284517.jpg (2011-10-15 12:02, 25.25 KB) / 该附件被下载次数 4
图片附件: 29284517.jpg (2011-10-15 12:02, 25.25 KB) / 该附件被下载次数 4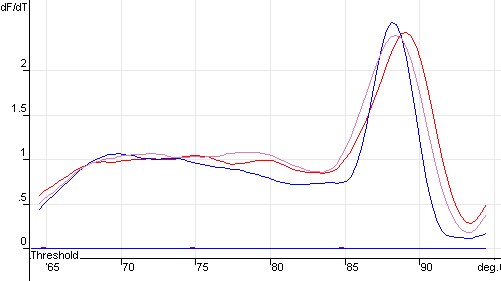 图片附件: 68967377.jpg (2011-10-15 12:03, 21.92 KB) / 该附件被下载次数 15
图片附件: 68967377.jpg (2011-10-15 12:03, 21.92 KB) / 该附件被下载次数 15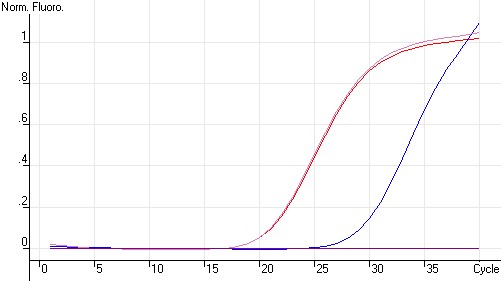 图片附件: 49910196_snap.jpg (2011-10-15 12:06, 74.64 KB) / 该附件被下载次数 5
图片附件: 49910196_snap.jpg (2011-10-15 12:06, 74.64 KB) / 该附件被下载次数 5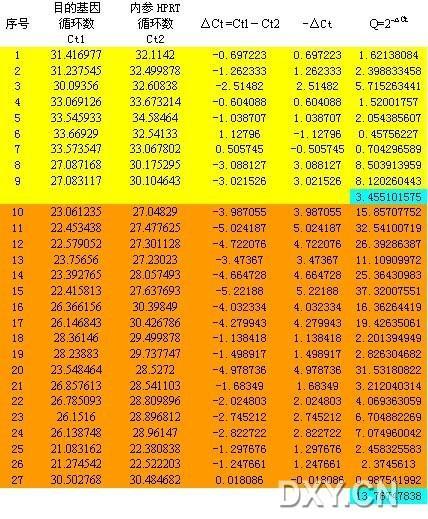 图片附件: 85807825.png (2011-10-15 12:09, 9.56 KB) / 该附件被下载次数 15
图片附件: 85807825.png (2011-10-15 12:09, 9.56 KB) / 该附件被下载次数 15 图片附件: 89321787_snap.jpg (2011-10-15 12:10, 36.32 KB) / 该附件被下载次数 10
图片附件: 89321787_snap.jpg (2011-10-15 12:10, 36.32 KB) / 该附件被下载次数 10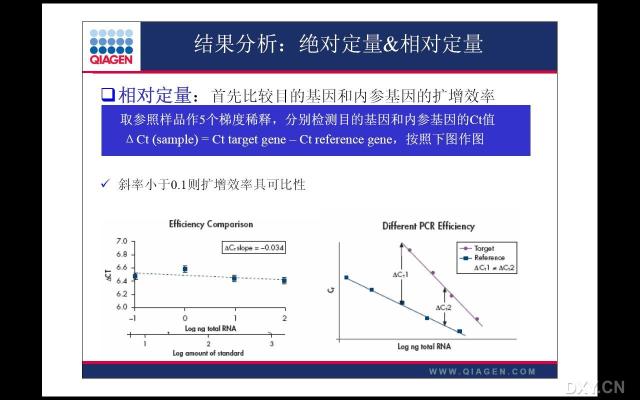 图片附件: 40970218_snap.jpg (2011-10-15 12:12, 21.79 KB) / 该附件被下载次数 3
图片附件: 40970218_snap.jpg (2011-10-15 12:12, 21.79 KB) / 该附件被下载次数 3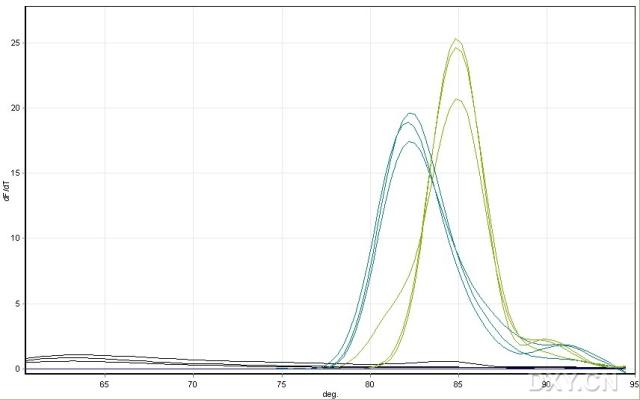 图片附件: 52569383.jpg (2011-10-15 12:17, 15.26 KB) / 该附件被下载次数 4
图片附件: 52569383.jpg (2011-10-15 12:17, 15.26 KB) / 该附件被下载次数 4 图片附件: 42847421_snap.jpg (2011-10-15 12:30, 41.76 KB) / 该附件被下载次数 7
图片附件: 42847421_snap.jpg (2011-10-15 12:30, 41.76 KB) / 该附件被下载次数 7 图片附件: 20992553.jpg (2011-10-15 12:31, 53.17 KB) / 该附件被下载次数 3
图片附件: 20992553.jpg (2011-10-15 12:31, 53.17 KB) / 该附件被下载次数 3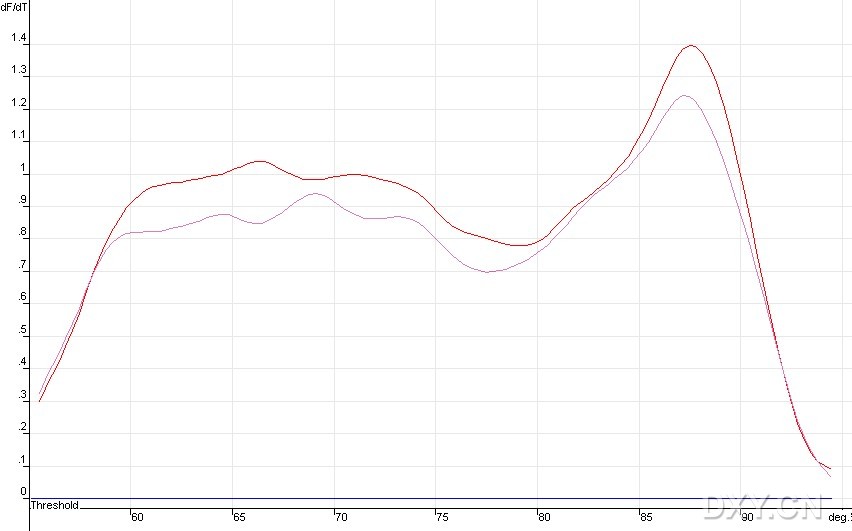 图片附件: 64391536.jpg (2011-10-15 12:31, 50.26 KB) / 该附件被下载次数 5
图片附件: 64391536.jpg (2011-10-15 12:31, 50.26 KB) / 该附件被下载次数 5 图片附件: 98947964.jpg (2011-10-15 12:38, 13.61 KB) / 该附件被下载次数 5
图片附件: 98947964.jpg (2011-10-15 12:38, 13.61 KB) / 该附件被下载次数 5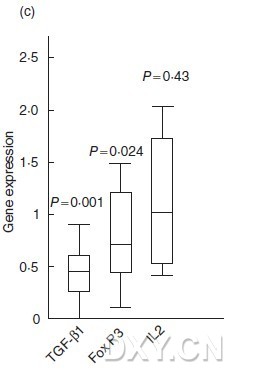 图片附件: 54275053.jpg (2011-10-15 12:41, 45.48 KB) / 该附件被下载次数 0
图片附件: 54275053.jpg (2011-10-15 12:41, 45.48 KB) / 该附件被下载次数 0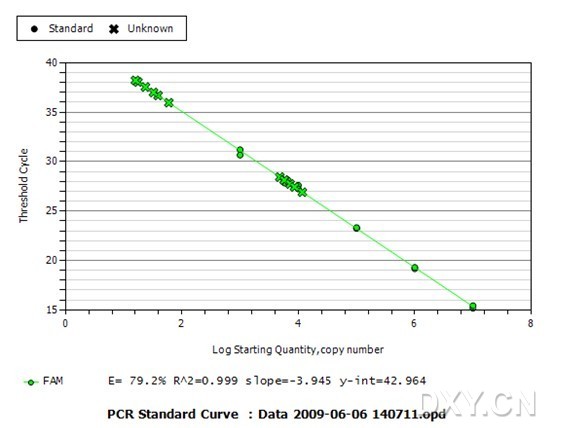 图片附件: 63327930_snap.jpg (2011-10-15 12:53, 42.29 KB) / 该附件被下载次数 2
图片附件: 63327930_snap.jpg (2011-10-15 12:53, 42.29 KB) / 该附件被下载次数 2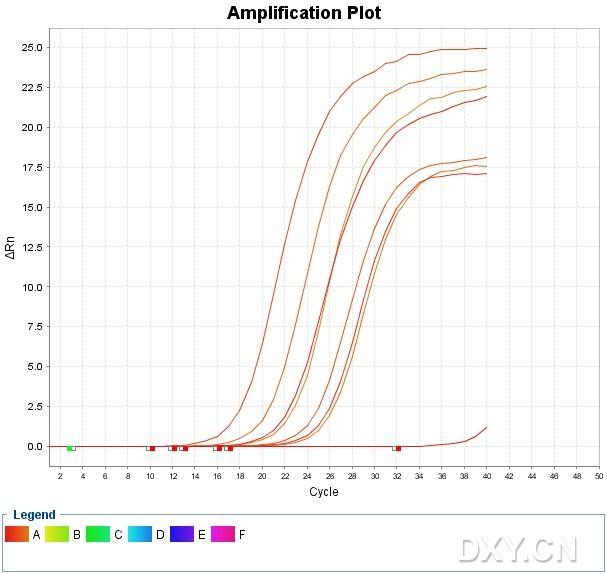 图片附件: 85894991_snap.jpg (2011-10-15 12:55, 74.48 KB) / 该附件被下载次数 14
图片附件: 85894991_snap.jpg (2011-10-15 12:55, 74.48 KB) / 该附件被下载次数 14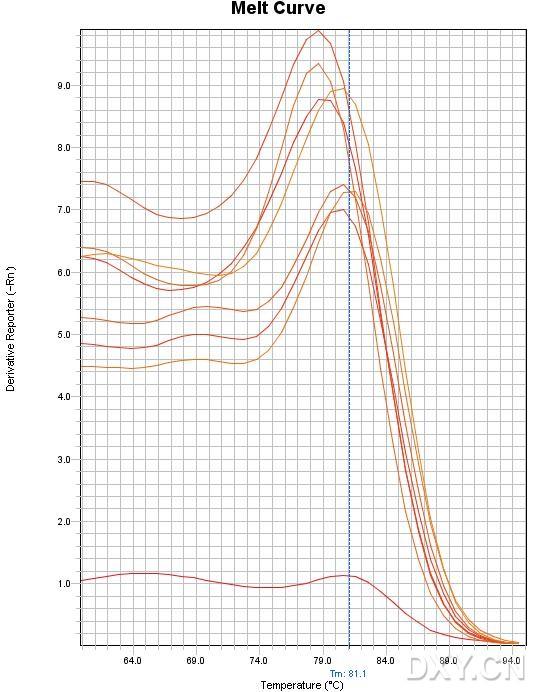 图片附件: 64355172.jpg (2011-10-15 13:00, 58.75 KB) / 该附件被下载次数 2
图片附件: 64355172.jpg (2011-10-15 13:00, 58.75 KB) / 该附件被下载次数 2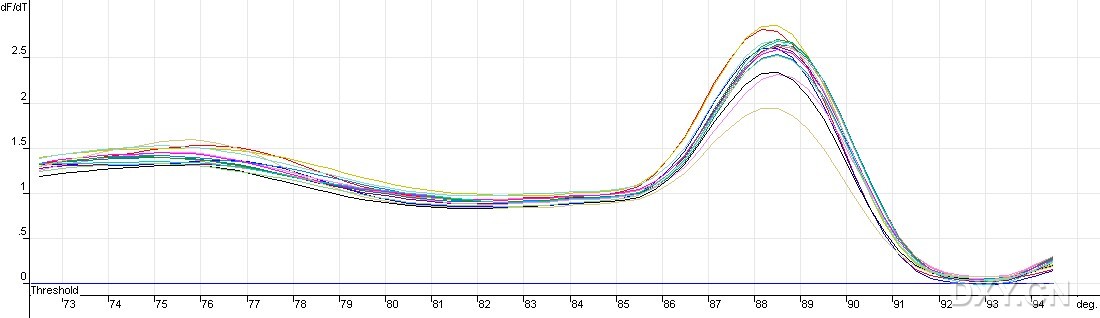 图片附件: 41182647_snap.jpg (2011-10-15 13:01, 15.25 KB) / 该附件被下载次数 4
图片附件: 41182647_snap.jpg (2011-10-15 13:01, 15.25 KB) / 该附件被下载次数 4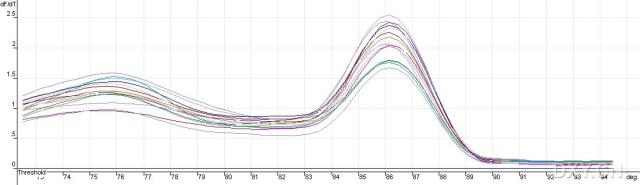 图片附件: 59796307_snap.jpg (2011-10-15 13:29, 19.76 KB) / 该附件被下载次数 10
图片附件: 59796307_snap.jpg (2011-10-15 13:29, 19.76 KB) / 该附件被下载次数 10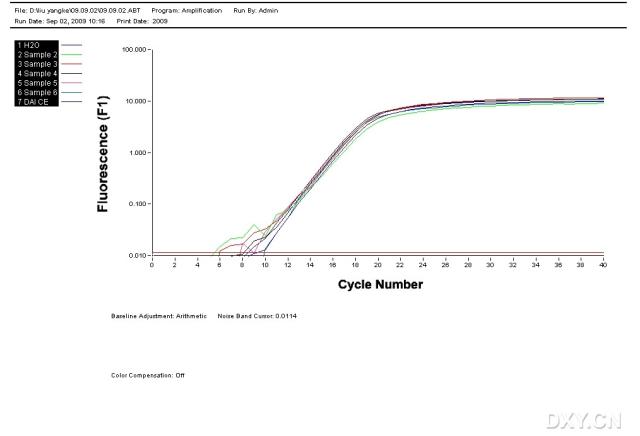 图片附件: 71215233.jpg (2011-10-15 13:40, 38.11 KB) / 该附件被下载次数 9
图片附件: 71215233.jpg (2011-10-15 13:40, 38.11 KB) / 该附件被下载次数 9 图片附件: 30204149.jpg (2011-10-15 13:41, 34.19 KB) / 该附件被下载次数 10
图片附件: 30204149.jpg (2011-10-15 13:41, 34.19 KB) / 该附件被下载次数 10 图片附件: 62814644.jpg (2011-10-15 13:42, 39.72 KB) / 该附件被下载次数 6
图片附件: 62814644.jpg (2011-10-15 13:42, 39.72 KB) / 该附件被下载次数 6 图片附件: 62814644.jpg (2011-10-15 13:44, 39.72 KB) / 该附件被下载次数 3
图片附件: 62814644.jpg (2011-10-15 13:44, 39.72 KB) / 该附件被下载次数 3 图片附件: 49715126.jpg (2011-10-15 13:45, 34.16 KB) / 该附件被下载次数 4
图片附件: 49715126.jpg (2011-10-15 13:45, 34.16 KB) / 该附件被下载次数 4 图片附件: 95440055.jpg (2011-10-15 13:46, 38.67 KB) / 该附件被下载次数 12
图片附件: 95440055.jpg (2011-10-15 13:46, 38.67 KB) / 该附件被下载次数 12 图片附件: 84163458_snap.jpg (2011-10-15 14:14, 60.74 KB) / 该附件被下载次数 6
图片附件: 84163458_snap.jpg (2011-10-15 14:14, 60.74 KB) / 该附件被下载次数 6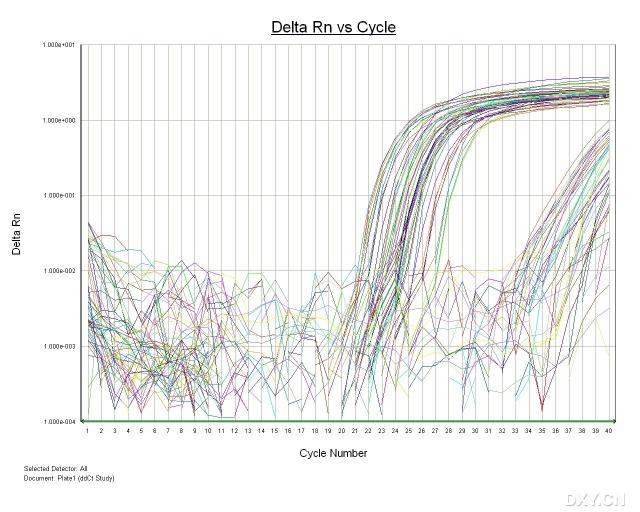 图片附件: 95974990.png (2011-10-15 15:18, 22.98 KB) / 该附件被下载次数 6
图片附件: 95974990.png (2011-10-15 15:18, 22.98 KB) / 该附件被下载次数 6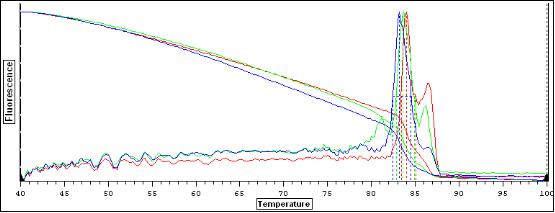 图片附件: 16807147.png (2011-10-15 15:18, 23.14 KB) / 该附件被下载次数 9
图片附件: 16807147.png (2011-10-15 15:18, 23.14 KB) / 该附件被下载次数 9 图片附件: 22753879_snap.jpg (2011-10-15 15:29, 22.15 KB) / 该附件被下载次数 2
图片附件: 22753879_snap.jpg (2011-10-15 15:29, 22.15 KB) / 该附件被下载次数 2 图片附件: 72171515.jpg (2011-10-16 13:15, 28.28 KB) / 该附件被下载次数 11
图片附件: 72171515.jpg (2011-10-16 13:15, 28.28 KB) / 该附件被下载次数 11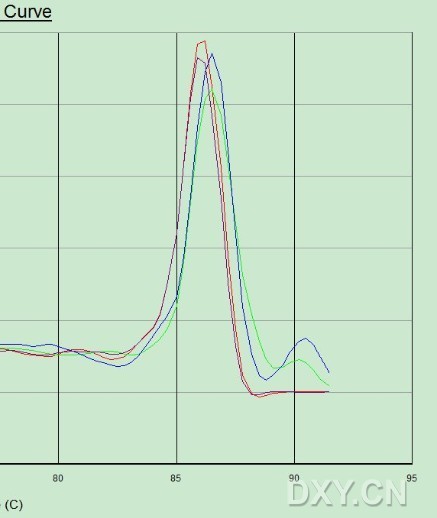 图片附件: 13811947.jpg (2011-10-16 13:17, 22.6 KB) / 该附件被下载次数 10
图片附件: 13811947.jpg (2011-10-16 13:17, 22.6 KB) / 该附件被下载次数 10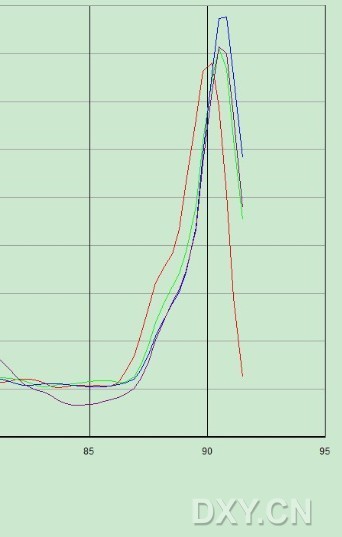 图片附件: 26247841_snap.jpg (2011-10-16 15:00, 36.54 KB) / 该附件被下载次数 7
图片附件: 26247841_snap.jpg (2011-10-16 15:00, 36.54 KB) / 该附件被下载次数 7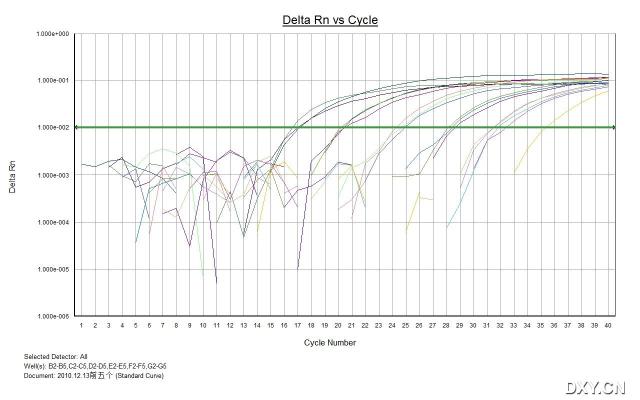 图片附件: 52929168_snap.jpg (2011-10-16 15:12, 40.05 KB) / 该附件被下载次数 4
图片附件: 52929168_snap.jpg (2011-10-16 15:12, 40.05 KB) / 该附件被下载次数 4 图片附件: 81068994.jpg (2011-10-16 15:17, 16.83 KB) / 该附件被下载次数 10
图片附件: 81068994.jpg (2011-10-16 15:17, 16.83 KB) / 该附件被下载次数 10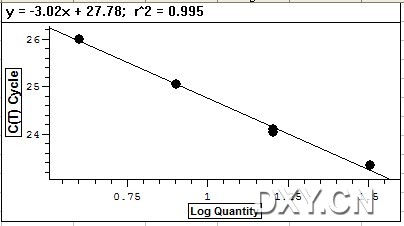 图片附件: 10800967.jpg (2011-10-16 15:34, 36.57 KB) / 该附件被下载次数 0
图片附件: 10800967.jpg (2011-10-16 15:34, 36.57 KB) / 该附件被下载次数 0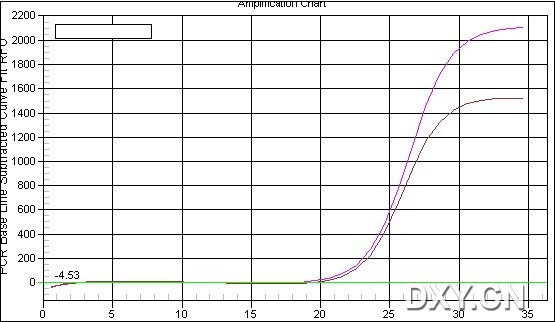 图片附件: 81142263.jpg (2011-10-16 15:34, 43.45 KB) / 该附件被下载次数 2
图片附件: 81142263.jpg (2011-10-16 15:34, 43.45 KB) / 该附件被下载次数 2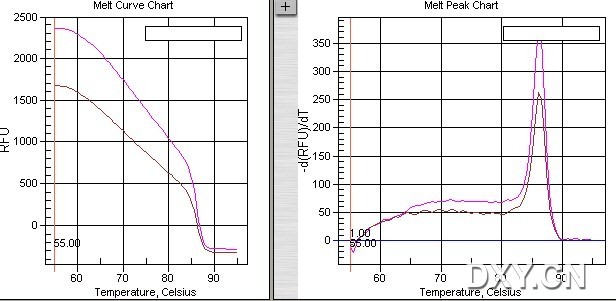
 图片附件: 52929168_snap.jpg (2011-10-16 15:47, 40.05 KB) / 该附件被下载次数 4
图片附件: 52929168_snap.jpg (2011-10-16 15:47, 40.05 KB) / 该附件被下载次数 4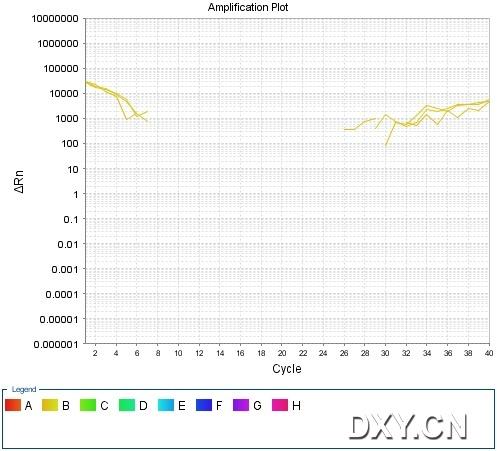 图片附件: 60405302.jpg (2011-10-16 15:50, 45.54 KB) / 该附件被下载次数 11
图片附件: 60405302.jpg (2011-10-16 15:50, 45.54 KB) / 该附件被下载次数 11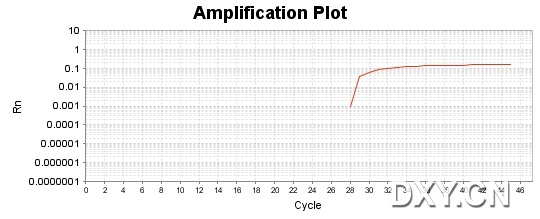 图片附件: 87638770.jpg (2011-10-16 15:51, 45.53 KB) / 该附件被下载次数 18
图片附件: 87638770.jpg (2011-10-16 15:51, 45.53 KB) / 该附件被下载次数 18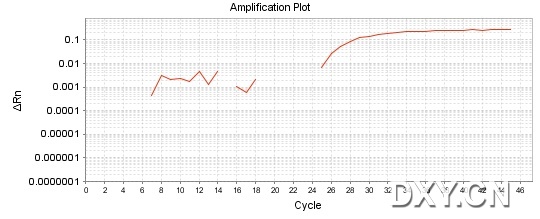 图片附件: 34075623.jpg (2011-10-16 15:51, 33.73 KB) / 该附件被下载次数 19
图片附件: 34075623.jpg (2011-10-16 15:51, 33.73 KB) / 该附件被下载次数 19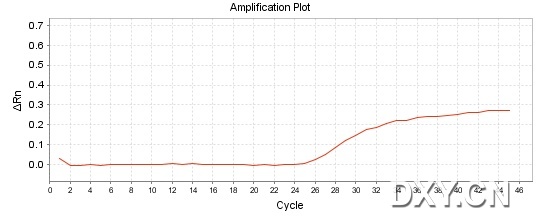 图片附件: 70836271.jpg (2011-10-16 15:51, 47.48 KB) / 该附件被下载次数 17
图片附件: 70836271.jpg (2011-10-16 15:51, 47.48 KB) / 该附件被下载次数 17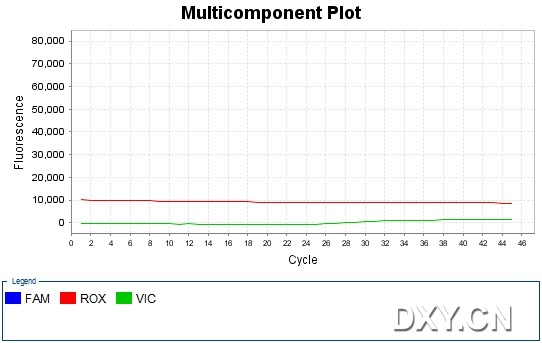 图片附件: 37358257.jpg (2011-10-16 15:52, 45.06 KB) / 该附件被下载次数 11
图片附件: 37358257.jpg (2011-10-16 15:52, 45.06 KB) / 该附件被下载次数 11 图片附件: 41078474.jpg (2011-10-16 15:53, 45.54 KB) / 该附件被下载次数 10
图片附件: 41078474.jpg (2011-10-16 15:53, 45.54 KB) / 该附件被下载次数 10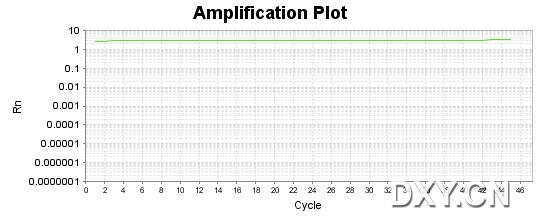 图片附件: 90667255.jpg (2011-10-16 15:53, 33.58 KB) / 该附件被下载次数 20
图片附件: 90667255.jpg (2011-10-16 15:53, 33.58 KB) / 该附件被下载次数 20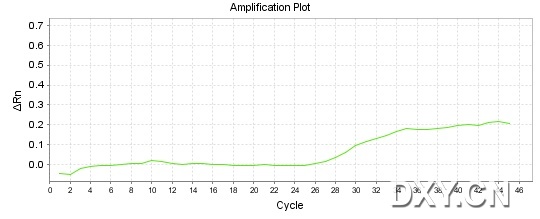 图片附件: 34268079.jpg (2011-10-16 15:53, 48.49 KB) / 该附件被下载次数 17
图片附件: 34268079.jpg (2011-10-16 15:53, 48.49 KB) / 该附件被下载次数 17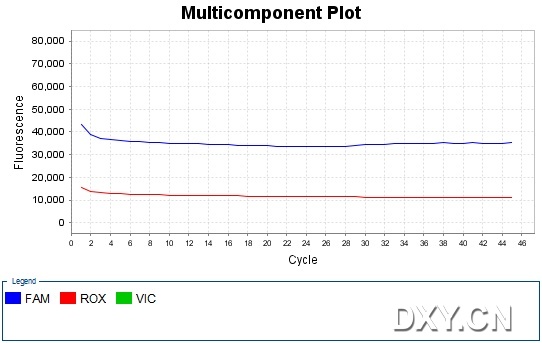
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |