
 图片附件: 91385623.jpg (2011-12-2 10:40, 32.61 KB) / 该附件被下载次数 7
图片附件: 91385623.jpg (2011-12-2 10:40, 32.61 KB) / 该附件被下载次数 7 图片附件: 36151209.snap.jpg (2011-12-2 10:46, 36.72 KB) / 该附件被下载次数 8
图片附件: 36151209.snap.jpg (2011-12-2 10:46, 36.72 KB) / 该附件被下载次数 8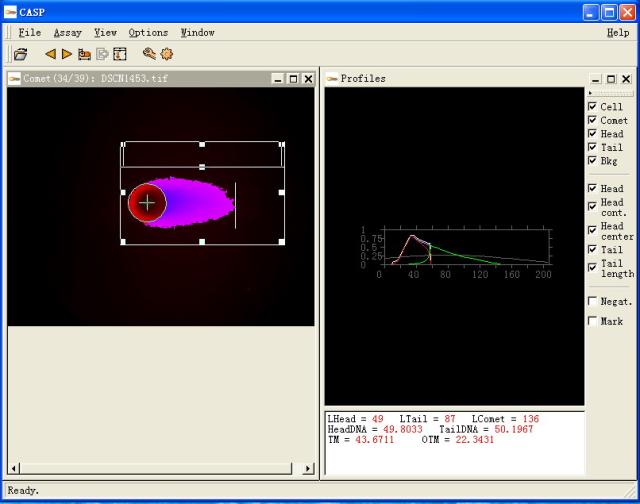 图片附件: 60525937.jpg (2011-12-2 11:14, 6.38 KB) / 该附件被下载次数 11
图片附件: 60525937.jpg (2011-12-2 11:14, 6.38 KB) / 该附件被下载次数 11 图片附件: 79494621.jpg (2011-12-2 11:15, 6.78 KB) / 该附件被下载次数 5
图片附件: 79494621.jpg (2011-12-2 11:15, 6.78 KB) / 该附件被下载次数 5 图片附件: 39363948.snap.jpg (2011-12-2 11:15, 29.9 KB) / 该附件被下载次数 7
图片附件: 39363948.snap.jpg (2011-12-2 11:15, 29.9 KB) / 该附件被下载次数 7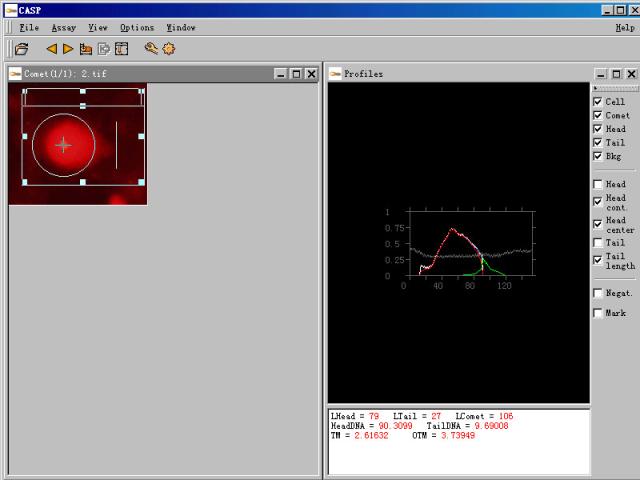 图片附件: 82205047.jpg (2011-12-2 11:30, 14.82 KB) / 该附件被下载次数 9
图片附件: 82205047.jpg (2011-12-2 11:30, 14.82 KB) / 该附件被下载次数 9 图片附件: 29025082.snap.jpg (2011-12-2 11:31, 36.32 KB) / 该附件被下载次数 6
图片附件: 29025082.snap.jpg (2011-12-2 11:31, 36.32 KB) / 该附件被下载次数 6 图片附件: 90280895.jpg (2011-12-2 11:31, 5.24 KB) / 该附件被下载次数 6
图片附件: 90280895.jpg (2011-12-2 11:31, 5.24 KB) / 该附件被下载次数 6 图片附件: 88795960.snap.jpg (2011-12-2 11:31, 29.47 KB) / 该附件被下载次数 7
图片附件: 88795960.snap.jpg (2011-12-2 11:31, 29.47 KB) / 该附件被下载次数 7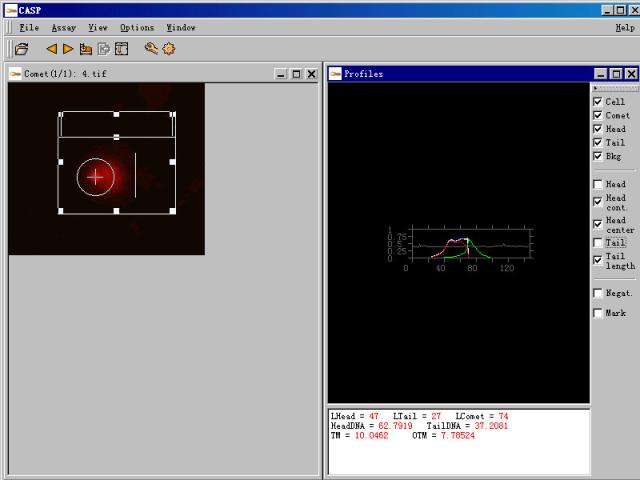 图片附件: 20243677.snap.jpg (2011-12-2 11:33, 6.25 KB) / 该附件被下载次数 5
图片附件: 20243677.snap.jpg (2011-12-2 11:33, 6.25 KB) / 该附件被下载次数 5 图片附件: 99808429.jpg (2011-12-2 11:36, 59.02 KB) / 该附件被下载次数 8
图片附件: 99808429.jpg (2011-12-2 11:36, 59.02 KB) / 该附件被下载次数 8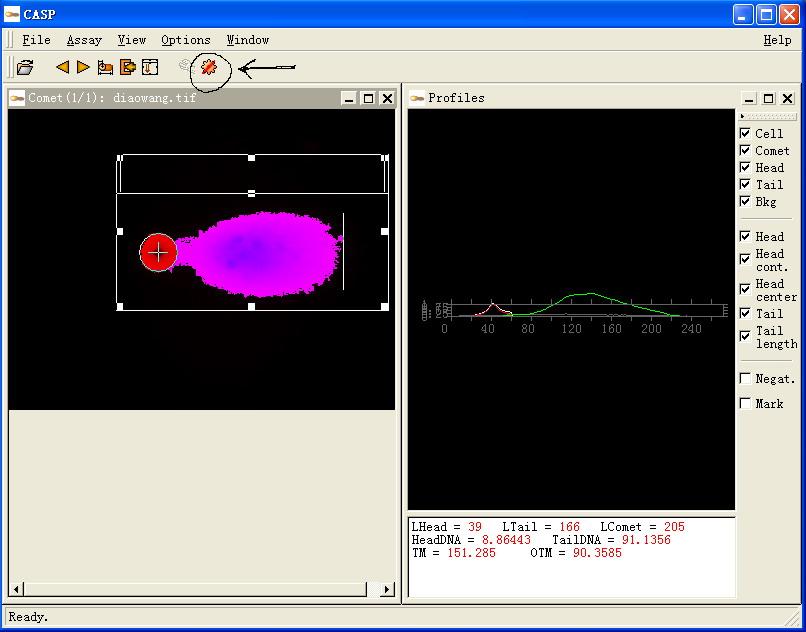 图片附件: 36898094.jpg (2011-12-2 11:41, 43.92 KB) / 该附件被下载次数 3
图片附件: 36898094.jpg (2011-12-2 11:41, 43.92 KB) / 该附件被下载次数 3 图片附件: 36898094.jpg (2011-12-2 11:44, 43.92 KB) / 该附件被下载次数 5
图片附件: 36898094.jpg (2011-12-2 11:44, 43.92 KB) / 该附件被下载次数 5 图片附件: 39373291.jpg (2011-12-2 11:44, 50.96 KB) / 该附件被下载次数 3
图片附件: 39373291.jpg (2011-12-2 11:44, 50.96 KB) / 该附件被下载次数 3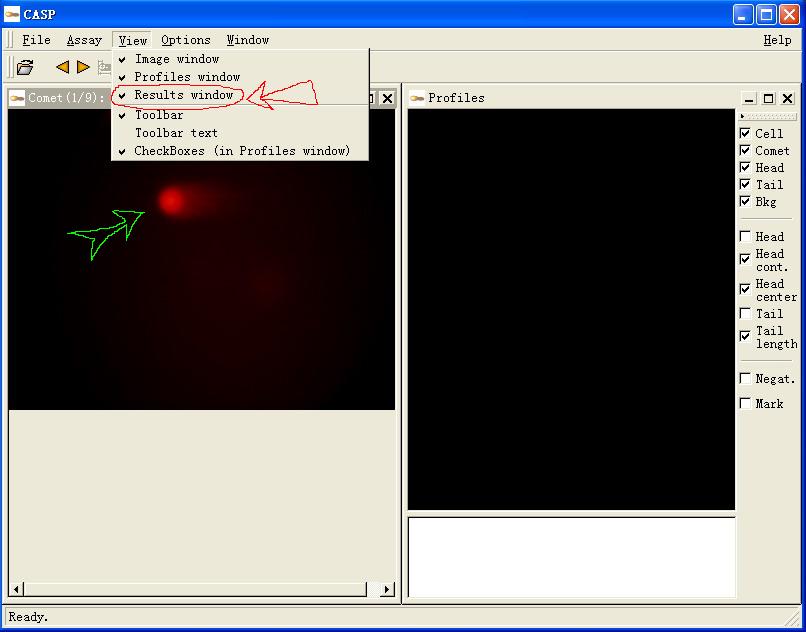 图片附件: 40748119.jpg (2011-12-2 11:44, 59.74 KB) / 该附件被下载次数 3
图片附件: 40748119.jpg (2011-12-2 11:44, 59.74 KB) / 该附件被下载次数 3 图片附件: 63527789.snap.jpg (2011-12-2 11:45, 49.44 KB) / 该附件被下载次数 7
图片附件: 63527789.snap.jpg (2011-12-2 11:45, 49.44 KB) / 该附件被下载次数 7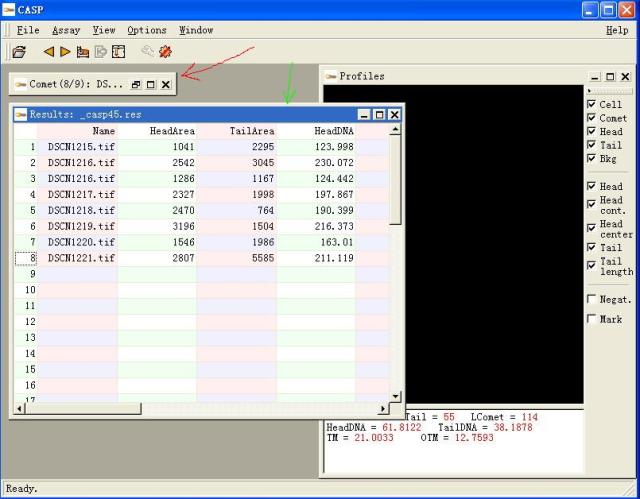 图片附件: 60969276.jpg (2011-12-2 11:45, 56.34 KB) / 该附件被下载次数 3
图片附件: 60969276.jpg (2011-12-2 11:45, 56.34 KB) / 该附件被下载次数 3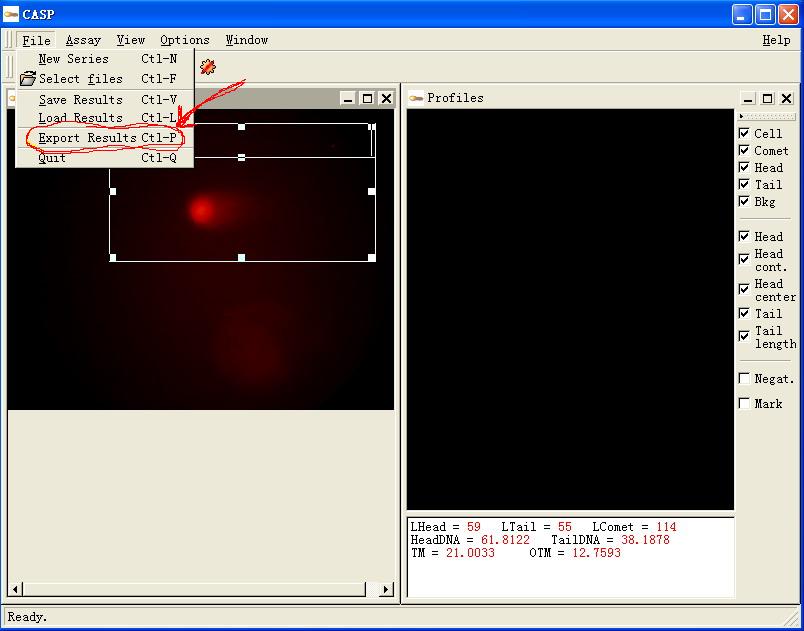 图片附件: 91591258.snap.jpg (2011-12-2 11:45, 42.78 KB) / 该附件被下载次数 5
图片附件: 91591258.snap.jpg (2011-12-2 11:45, 42.78 KB) / 该附件被下载次数 5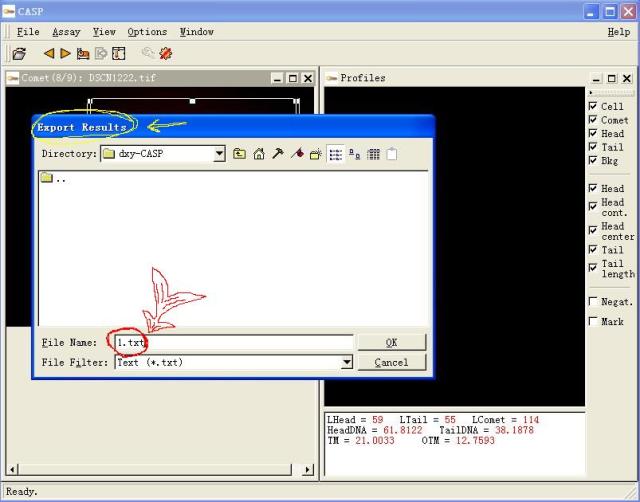 图片附件: 63947964.snap.jpg (2011-12-2 11:46, 64.08 KB) / 该附件被下载次数 5
图片附件: 63947964.snap.jpg (2011-12-2 11:46, 64.08 KB) / 该附件被下载次数 5 图片附件: 44511746.snap.jpg (2011-12-2 11:46, 63.01 KB) / 该附件被下载次数 4
图片附件: 44511746.snap.jpg (2011-12-2 11:46, 63.01 KB) / 该附件被下载次数 4 图片附件: 75889937.jpg (2011-12-2 11:48, 9.59 KB) / 该附件被下载次数 5
图片附件: 75889937.jpg (2011-12-2 11:48, 9.59 KB) / 该附件被下载次数 5 图片附件: 60969779.jpg (2011-12-2 11:48, 24.03 KB) / 该附件被下载次数 3
图片附件: 60969779.jpg (2011-12-2 11:48, 24.03 KB) / 该附件被下载次数 3 图片附件: 45727309.jpg (2011-12-2 11:49, 7.84 KB) / 该附件被下载次数 8
图片附件: 45727309.jpg (2011-12-2 11:49, 7.84 KB) / 该附件被下载次数 8 图片附件: 21800678.jpg (2011-12-2 11:49, 6.3 KB) / 该附件被下载次数 6
图片附件: 21800678.jpg (2011-12-2 11:49, 6.3 KB) / 该附件被下载次数 6 图片附件: 54491725.jpg (2011-12-2 11:49, 10.38 KB) / 该附件被下载次数 7
图片附件: 54491725.jpg (2011-12-2 11:49, 10.38 KB) / 该附件被下载次数 7 图片附件: 18099370.snap.jpg (2011-12-2 11:50, 29.47 KB) / 该附件被下载次数 3
图片附件: 18099370.snap.jpg (2011-12-2 11:50, 29.47 KB) / 该附件被下载次数 3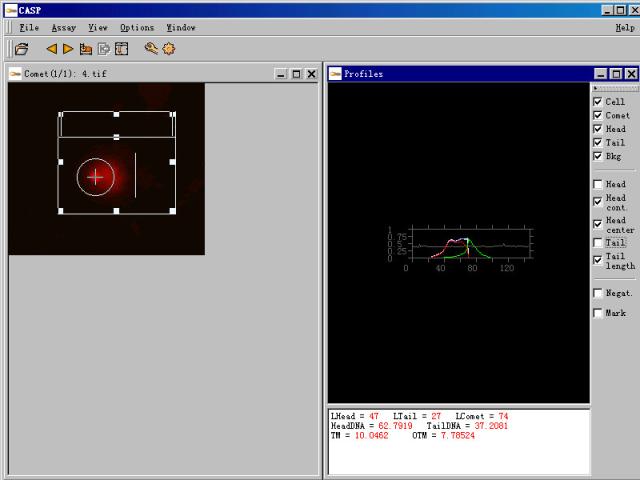 图片附件: 54805903.snap.jpg (2011-12-2 14:54, 25.37 KB) / 该附件被下载次数 1
图片附件: 54805903.snap.jpg (2011-12-2 14:54, 25.37 KB) / 该附件被下载次数 1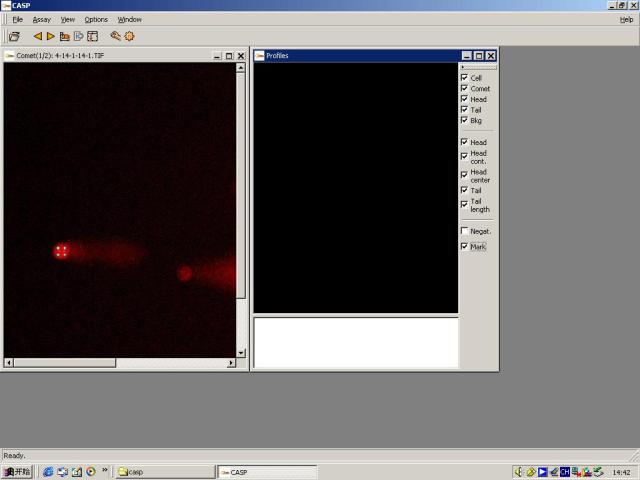 图片附件: 87087887.snap.jpg (2011-12-2 15:33, 9.4 KB) / 该附件被下载次数 6
图片附件: 87087887.snap.jpg (2011-12-2 15:33, 9.4 KB) / 该附件被下载次数 6 图片附件: 78729516.snap.jpg (2011-12-2 15:33, 5.91 KB) / 该附件被下载次数 15
图片附件: 78729516.snap.jpg (2011-12-2 15:33, 5.91 KB) / 该附件被下载次数 15 图片附件: 18228065.snap.jpg (2011-12-2 15:34, 27.47 KB) / 该附件被下载次数 9
图片附件: 18228065.snap.jpg (2011-12-2 15:34, 27.47 KB) / 该附件被下载次数 9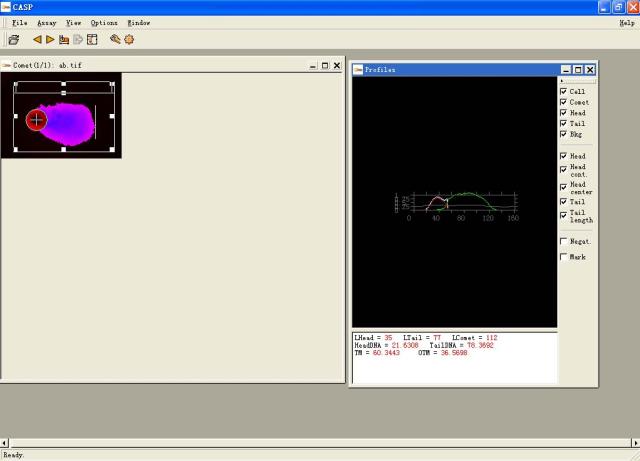 图片附件: 83168672.jpg (2011-12-2 15:57, 9.33 KB) / 该附件被下载次数 2
图片附件: 83168672.jpg (2011-12-2 15:57, 9.33 KB) / 该附件被下载次数 2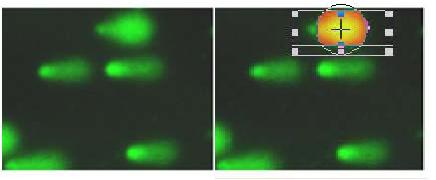 图片附件: 93387816.snap.jpg (2011-12-2 15:58, 7.72 KB) / 该附件被下载次数 3
图片附件: 93387816.snap.jpg (2011-12-2 15:58, 7.72 KB) / 该附件被下载次数 3 图片附件: 65045811.jpg (2011-12-2 16:04, 28.05 KB) / 该附件被下载次数 5
图片附件: 65045811.jpg (2011-12-2 16:04, 28.05 KB) / 该附件被下载次数 5 图片附件: 74249448.jpg (2011-12-2 16:14, 23.2 KB) / 该附件被下载次数 2
图片附件: 74249448.jpg (2011-12-2 16:14, 23.2 KB) / 该附件被下载次数 2 图片附件: 80520519.jpg (2011-12-2 16:19, 52.03 KB) / 该附件被下载次数 1
图片附件: 80520519.jpg (2011-12-2 16:19, 52.03 KB) / 该附件被下载次数 1 图片附件: 95274879.jpg (2011-12-2 16:20, 41.97 KB) / 该附件被下载次数 0
图片附件: 95274879.jpg (2011-12-2 16:20, 41.97 KB) / 该附件被下载次数 0 图片附件: 17511211.jpg (2011-12-2 16:39, 22.29 KB) / 该附件被下载次数 0
图片附件: 17511211.jpg (2011-12-2 16:39, 22.29 KB) / 该附件被下载次数 0 图片附件: 93296500.snap.jpg (2011-12-2 16:42, 9.36 KB) / 该附件被下载次数 0
图片附件: 93296500.snap.jpg (2011-12-2 16:42, 9.36 KB) / 该附件被下载次数 0 图片附件: 16638139.jpg (2011-12-2 17:02, 1.55 KB) / 该附件被下载次数 1
图片附件: 16638139.jpg (2011-12-2 17:02, 1.55 KB) / 该附件被下载次数 1 图片附件: 77102731.jpg (2011-12-2 17:03, 8.52 KB) / 该附件被下载次数 4
图片附件: 77102731.jpg (2011-12-2 17:03, 8.52 KB) / 该附件被下载次数 4 图片附件: 42268245.jpg (2011-12-2 17:24, 7.94 KB) / 该附件被下载次数 1
图片附件: 42268245.jpg (2011-12-2 17:24, 7.94 KB) / 该附件被下载次数 1 图片附件: 34271692.jpg (2011-12-2 17:25, 40.57 KB) / 该附件被下载次数 1
图片附件: 34271692.jpg (2011-12-2 17:25, 40.57 KB) / 该附件被下载次数 1 图片附件: 89922028.snap.jpg (2011-12-2 17:25, 10.24 KB) / 该附件被下载次数 0
图片附件: 89922028.snap.jpg (2011-12-2 17:25, 10.24 KB) / 该附件被下载次数 0 图片附件: 61171110.jpg (2011-12-2 17:29, 14.77 KB) / 该附件被下载次数 2
图片附件: 61171110.jpg (2011-12-2 17:29, 14.77 KB) / 该附件被下载次数 2 图片附件: 46014350.jpg (2011-12-2 17:30, 56.51 KB) / 该附件被下载次数 1
图片附件: 46014350.jpg (2011-12-2 17:30, 56.51 KB) / 该附件被下载次数 1 图片附件: 91206664.jpg (2011-12-2 17:34, 3.74 KB) / 该附件被下载次数 1
图片附件: 91206664.jpg (2011-12-2 17:34, 3.74 KB) / 该附件被下载次数 1 图片附件: 52261331.jpg (2011-12-3 09:01, 20.69 KB) / 该附件被下载次数 2
图片附件: 52261331.jpg (2011-12-3 09:01, 20.69 KB) / 该附件被下载次数 2 图片附件: 73531546.snap.jpg (2011-12-3 09:27, 51.19 KB) / 该附件被下载次数 4
图片附件: 73531546.snap.jpg (2011-12-3 09:27, 51.19 KB) / 该附件被下载次数 4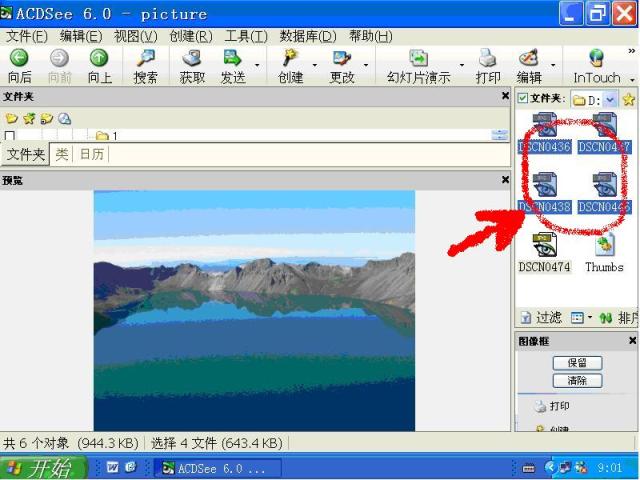 图片附件: 82593645.snap.jpg (2011-12-3 09:27, 63.84 KB) / 该附件被下载次数 2
图片附件: 82593645.snap.jpg (2011-12-3 09:27, 63.84 KB) / 该附件被下载次数 2 图片附件: 77548859.snap.jpg (2011-12-3 09:27, 60.92 KB) / 该附件被下载次数 2
图片附件: 77548859.snap.jpg (2011-12-3 09:27, 60.92 KB) / 该附件被下载次数 2 图片附件: 14723710.snap.jpg (2011-12-3 09:28, 56.19 KB) / 该附件被下载次数 2
图片附件: 14723710.snap.jpg (2011-12-3 09:28, 56.19 KB) / 该附件被下载次数 2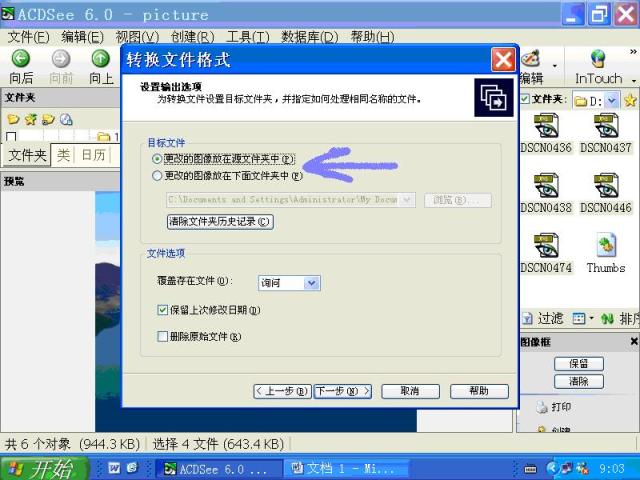 图片附件: 35290419.snap.jpg (2011-12-3 09:28, 59.12 KB) / 该附件被下载次数 3
图片附件: 35290419.snap.jpg (2011-12-3 09:28, 59.12 KB) / 该附件被下载次数 3 图片附件: 45939867.snap.jpg (2011-12-3 09:29, 53.14 KB) / 该附件被下载次数 4
图片附件: 45939867.snap.jpg (2011-12-3 09:29, 53.14 KB) / 该附件被下载次数 4 图片附件: 87263868.snap.jpg (2011-12-3 09:29, 53.77 KB) / 该附件被下载次数 1
图片附件: 87263868.snap.jpg (2011-12-3 09:29, 53.77 KB) / 该附件被下载次数 1 图片附件: 31284792.snap.jpg (2011-12-3 09:30, 64.75 KB) / 该附件被下载次数 4
图片附件: 31284792.snap.jpg (2011-12-3 09:30, 64.75 KB) / 该附件被下载次数 4 图片附件: 20483910.snap.jpg (2011-12-3 09:30, 54.46 KB) / 该附件被下载次数 2
图片附件: 20483910.snap.jpg (2011-12-3 09:30, 54.46 KB) / 该附件被下载次数 2 图片附件: 88826200.jpg (2011-12-3 14:34, 2.06 KB) / 该附件被下载次数 5
图片附件: 88826200.jpg (2011-12-3 14:34, 2.06 KB) / 该附件被下载次数 5 图片附件: 74606060.jpg (2011-12-3 14:35, 34.44 KB) / 该附件被下载次数 4
图片附件: 74606060.jpg (2011-12-3 14:35, 34.44 KB) / 该附件被下载次数 4 图片附件: 74110127.jpg (2011-12-3 14:42, 48.18 KB) / 该附件被下载次数 2
图片附件: 74110127.jpg (2011-12-3 14:42, 48.18 KB) / 该附件被下载次数 2 图片附件: 86879251.jpg (2011-12-3 14:50, 20.95 KB) / 该附件被下载次数 1
图片附件: 86879251.jpg (2011-12-3 14:50, 20.95 KB) / 该附件被下载次数 1 图片附件: 87242310.snap.jpg (2011-12-3 16:02, 34.56 KB) / 该附件被下载次数 2
图片附件: 87242310.snap.jpg (2011-12-3 16:02, 34.56 KB) / 该附件被下载次数 2 图片附件: 54368783.snap.jpg (2011-12-3 16:11, 37.27 KB) / 该附件被下载次数 3
图片附件: 54368783.snap.jpg (2011-12-3 16:11, 37.27 KB) / 该附件被下载次数 3 图片附件: 41352639.snap.jpg (2011-12-4 14:39, 8.51 KB) / 该附件被下载次数 0
图片附件: 41352639.snap.jpg (2011-12-4 14:39, 8.51 KB) / 该附件被下载次数 0 图片附件: 21063874.jpg (2011-12-4 15:45, 19.23 KB) / 该附件被下载次数 4
图片附件: 21063874.jpg (2011-12-4 15:45, 19.23 KB) / 该附件被下载次数 4 图片附件: 25546426.snap.jpg (2011-12-4 15:47, 12.28 KB) / 该附件被下载次数 3
图片附件: 25546426.snap.jpg (2011-12-4 15:47, 12.28 KB) / 该附件被下载次数 3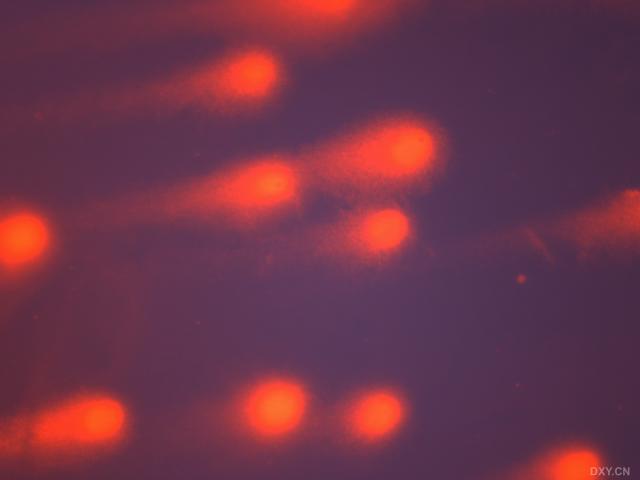 图片附件: 96277002.snap.jpg (2011-12-4 15:47, 11.33 KB) / 该附件被下载次数 3
图片附件: 96277002.snap.jpg (2011-12-4 15:47, 11.33 KB) / 该附件被下载次数 3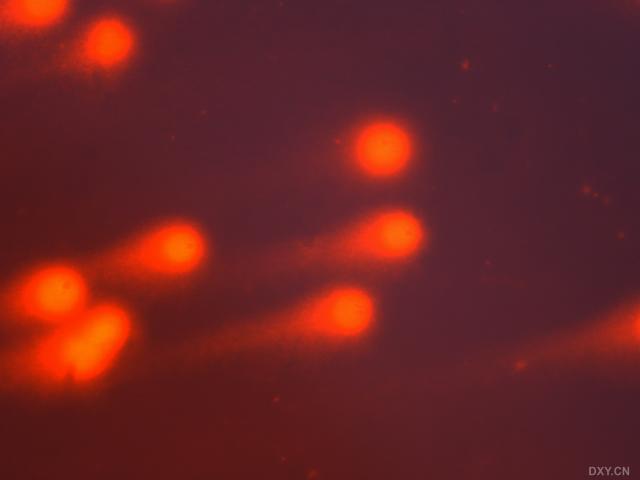 图片附件: 13575172.jpg (2011-12-4 15:48, 17.02 KB) / 该附件被下载次数 3
图片附件: 13575172.jpg (2011-12-4 15:48, 17.02 KB) / 该附件被下载次数 3 图片附件: 54664398.jpg (2011-12-4 15:55, 13.34 KB) / 该附件被下载次数 3
图片附件: 54664398.jpg (2011-12-4 15:55, 13.34 KB) / 该附件被下载次数 3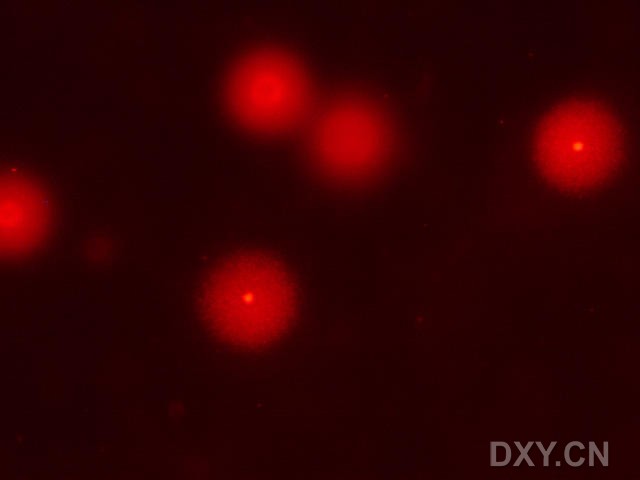 图片附件: 64278276.jpg (2011-12-4 15:56, 10.05 KB) / 该附件被下载次数 3
图片附件: 64278276.jpg (2011-12-4 15:56, 10.05 KB) / 该附件被下载次数 3 图片附件: 11771013.jpg (2011-12-4 15:56, 10.91 KB) / 该附件被下载次数 7
图片附件: 11771013.jpg (2011-12-4 15:56, 10.91 KB) / 该附件被下载次数 7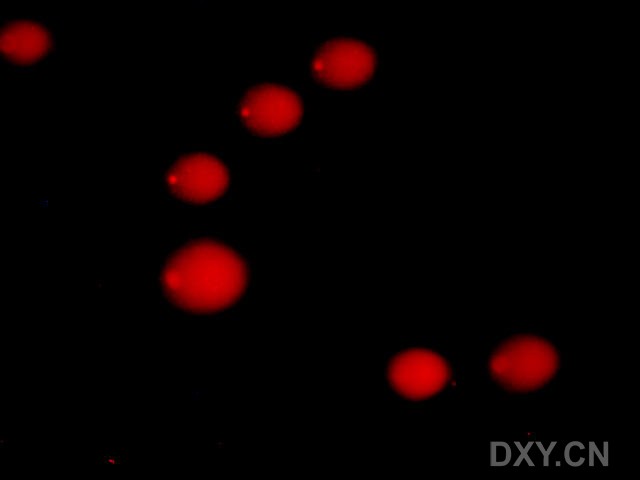 图片附件: 67901876.jpg (2011-12-4 15:57, 13.53 KB) / 该附件被下载次数 3
图片附件: 67901876.jpg (2011-12-4 15:57, 13.53 KB) / 该附件被下载次数 3 图片附件: 94142866.jpg (2011-12-4 15:57, 10.62 KB) / 该附件被下载次数 2
图片附件: 94142866.jpg (2011-12-4 15:57, 10.62 KB) / 该附件被下载次数 2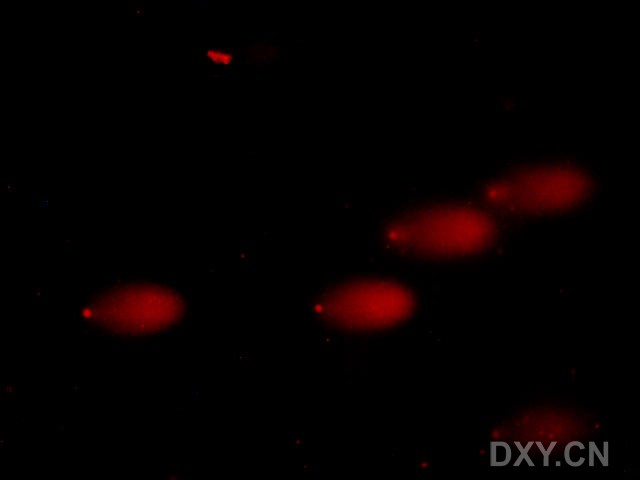 图片附件: 70375308.jpg (2011-12-4 15:58, 8.37 KB) / 该附件被下载次数 6
图片附件: 70375308.jpg (2011-12-4 15:58, 8.37 KB) / 该附件被下载次数 6 图片附件: 23401379.jpg (2011-12-4 16:04, 17.83 KB) / 该附件被下载次数 3
图片附件: 23401379.jpg (2011-12-4 16:04, 17.83 KB) / 该附件被下载次数 3 图片附件: 35339674.jpg (2011-12-4 16:05, 39.22 KB) / 该附件被下载次数 2
图片附件: 35339674.jpg (2011-12-4 16:05, 39.22 KB) / 该附件被下载次数 2 图片附件: 30171573.jpg (2011-12-4 16:08, 8.94 KB) / 该附件被下载次数 1
图片附件: 30171573.jpg (2011-12-4 16:08, 8.94 KB) / 该附件被下载次数 1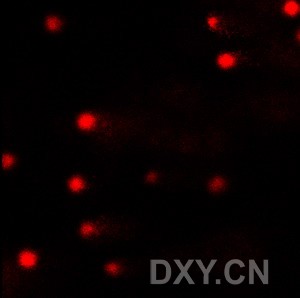 图片附件: 96126024.jpg (2011-12-4 16:08, 9.74 KB) / 该附件被下载次数 2
图片附件: 96126024.jpg (2011-12-4 16:08, 9.74 KB) / 该附件被下载次数 2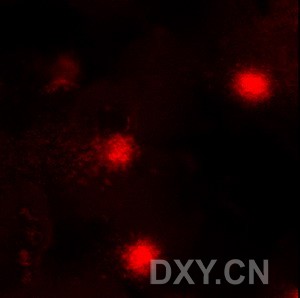 图片附件: 30563004.jpg (2011-12-4 16:13, 51.19 KB) / 该附件被下载次数 5
图片附件: 30563004.jpg (2011-12-4 16:13, 51.19 KB) / 该附件被下载次数 5 图片附件: 28968849.snap (1).jpg (2011-12-4 16:26, 32.87 KB) / 该附件被下载次数 3
图片附件: 28968849.snap (1).jpg (2011-12-4 16:26, 32.87 KB) / 该附件被下载次数 3 图片附件: 19928314.jpg (2011-12-4 16:27, 9.81 KB) / 该附件被下载次数 6
图片附件: 19928314.jpg (2011-12-4 16:27, 9.81 KB) / 该附件被下载次数 6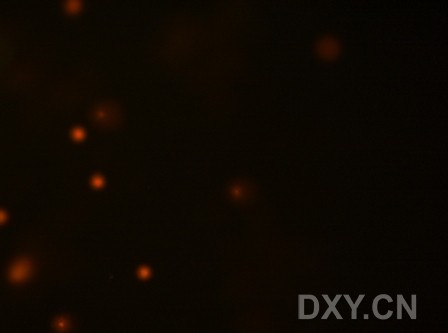 图片附件: 82078432.jpg (2011-12-4 16:45, 7.46 KB) / 该附件被下载次数 0
图片附件: 82078432.jpg (2011-12-4 16:45, 7.46 KB) / 该附件被下载次数 0 图片附件: 40129212.jpg (2011-12-4 16:45, 8.02 KB) / 该附件被下载次数 0
图片附件: 40129212.jpg (2011-12-4 16:45, 8.02 KB) / 该附件被下载次数 0 图片附件: 86352134.jpg (2011-12-4 16:55, 7.49 KB) / 该附件被下载次数 4
图片附件: 86352134.jpg (2011-12-4 16:55, 7.49 KB) / 该附件被下载次数 4 图片附件: 76911514.snap.jpg (2011-12-5 11:02, 19.11 KB) / 该附件被下载次数 4
图片附件: 76911514.snap.jpg (2011-12-5 11:02, 19.11 KB) / 该附件被下载次数 4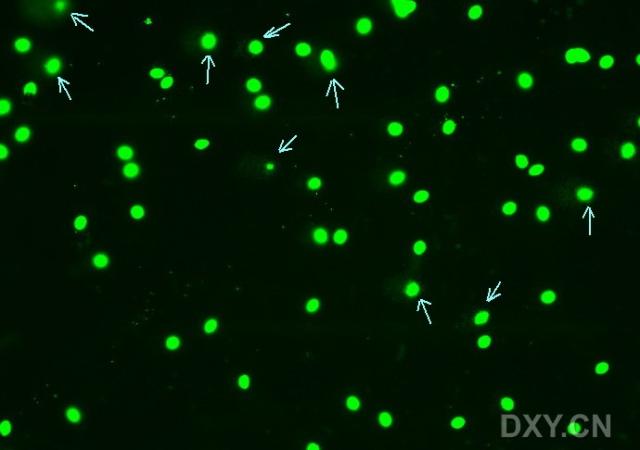 图片附件: 62338143.jpg (2011-12-5 11:12, 30.86 KB) / 该附件被下载次数 5
图片附件: 62338143.jpg (2011-12-5 11:12, 30.86 KB) / 该附件被下载次数 5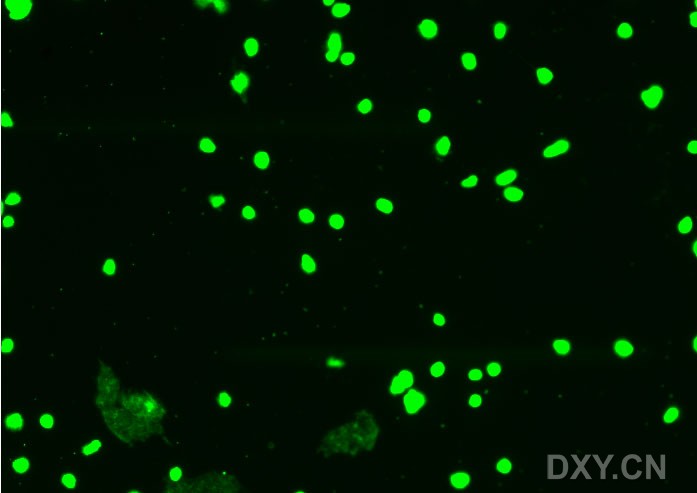 图片附件: 62468415.snap.jpg (2011-12-5 14:47, 42.63 KB) / 该附件被下载次数 4
图片附件: 62468415.snap.jpg (2011-12-5 14:47, 42.63 KB) / 该附件被下载次数 4 图片附件: 27029486.jpg (2011-12-5 15:04, 17.42 KB) / 该附件被下载次数 5
图片附件: 27029486.jpg (2011-12-5 15:04, 17.42 KB) / 该附件被下载次数 5 图片附件: 48723879.jpg (2011-12-5 15:04, 28.73 KB) / 该附件被下载次数 4
图片附件: 48723879.jpg (2011-12-5 15:04, 28.73 KB) / 该附件被下载次数 4 图片附件: 63448829.jpg (2011-12-6 09:58, 25.66 KB) / 该附件被下载次数 1
图片附件: 63448829.jpg (2011-12-6 09:58, 25.66 KB) / 该附件被下载次数 1 图片附件: 34627466.jpg (2011-12-6 09:59, 25.21 KB) / 该附件被下载次数 5
图片附件: 34627466.jpg (2011-12-6 09:59, 25.21 KB) / 该附件被下载次数 5 图片附件: 95601221.jpg (2011-12-6 10:20, 24.63 KB) / 该附件被下载次数 4
图片附件: 95601221.jpg (2011-12-6 10:20, 24.63 KB) / 该附件被下载次数 4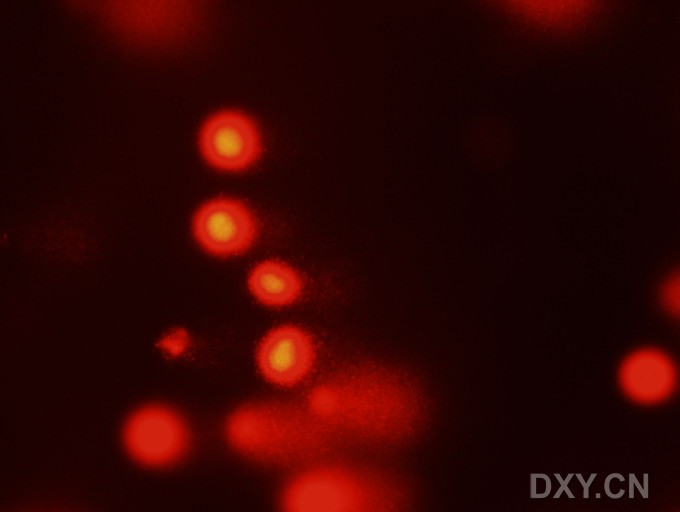 图片附件: 92279606.jpg (2011-12-6 10:57, 40.75 KB) / 该附件被下载次数 2
图片附件: 92279606.jpg (2011-12-6 10:57, 40.75 KB) / 该附件被下载次数 2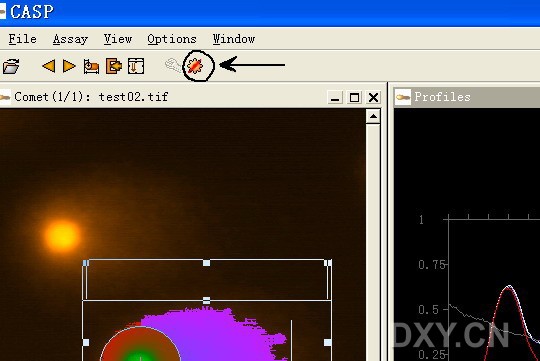 图片附件: 41324128.snap.jpg (2011-12-6 11:37, 65.32 KB) / 该附件被下载次数 4
图片附件: 41324128.snap.jpg (2011-12-6 11:37, 65.32 KB) / 该附件被下载次数 4 图片附件: 90071880.snap.jpg (2011-12-6 11:38, 64 KB) / 该附件被下载次数 5
图片附件: 90071880.snap.jpg (2011-12-6 11:38, 64 KB) / 该附件被下载次数 5 图片附件: 55664244.snap.jpg (2011-12-6 11:38, 43.78 KB) / 该附件被下载次数 3
图片附件: 55664244.snap.jpg (2011-12-6 11:38, 43.78 KB) / 该附件被下载次数 3 图片附件: 47411883.snap.jpg (2011-12-6 11:39, 61.39 KB) / 该附件被下载次数 1
图片附件: 47411883.snap.jpg (2011-12-6 11:39, 61.39 KB) / 该附件被下载次数 1 图片附件: 64448097.jpg (2011-12-6 14:45, 19.6 KB) / 该附件被下载次数 4
图片附件: 64448097.jpg (2011-12-6 14:45, 19.6 KB) / 该附件被下载次数 4 图片附件: 57599314.jpg (2011-12-6 15:05, 21.91 KB) / 该附件被下载次数 5
图片附件: 57599314.jpg (2011-12-6 15:05, 21.91 KB) / 该附件被下载次数 5 图片附件: 95601221.jpg (2011-12-6 15:05, 24.63 KB) / 该附件被下载次数 4
图片附件: 95601221.jpg (2011-12-6 15:05, 24.63 KB) / 该附件被下载次数 4 图片附件: 37742221.jpg (2011-12-6 15:06, 22.86 KB) / 该附件被下载次数 3
图片附件: 37742221.jpg (2011-12-6 15:06, 22.86 KB) / 该附件被下载次数 3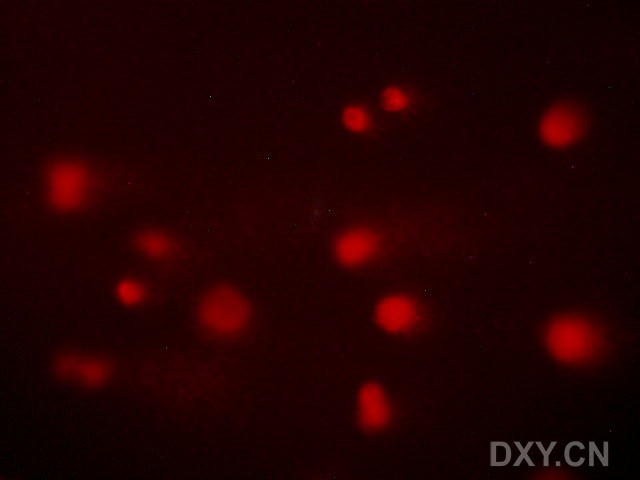 图片附件: 57386591.jpg (2011-12-6 15:44, 78.03 KB) / 该附件被下载次数 3
图片附件: 57386591.jpg (2011-12-6 15:44, 78.03 KB) / 该附件被下载次数 3 图片附件: 17409386.jpg (2011-12-6 15:51, 6.26 KB) / 该附件被下载次数 3
图片附件: 17409386.jpg (2011-12-6 15:51, 6.26 KB) / 该附件被下载次数 3 图片附件: 61432954.jpg (2011-12-6 16:25, 15.38 KB) / 该附件被下载次数 3
图片附件: 61432954.jpg (2011-12-6 16:25, 15.38 KB) / 该附件被下载次数 3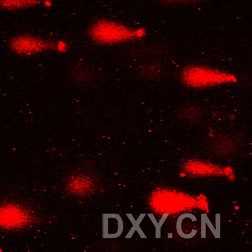 图片附件: 33871733.jpg (2011-12-6 16:49, 116.13 KB) / 该附件被下载次数 1
图片附件: 33871733.jpg (2011-12-6 16:49, 116.13 KB) / 该附件被下载次数 1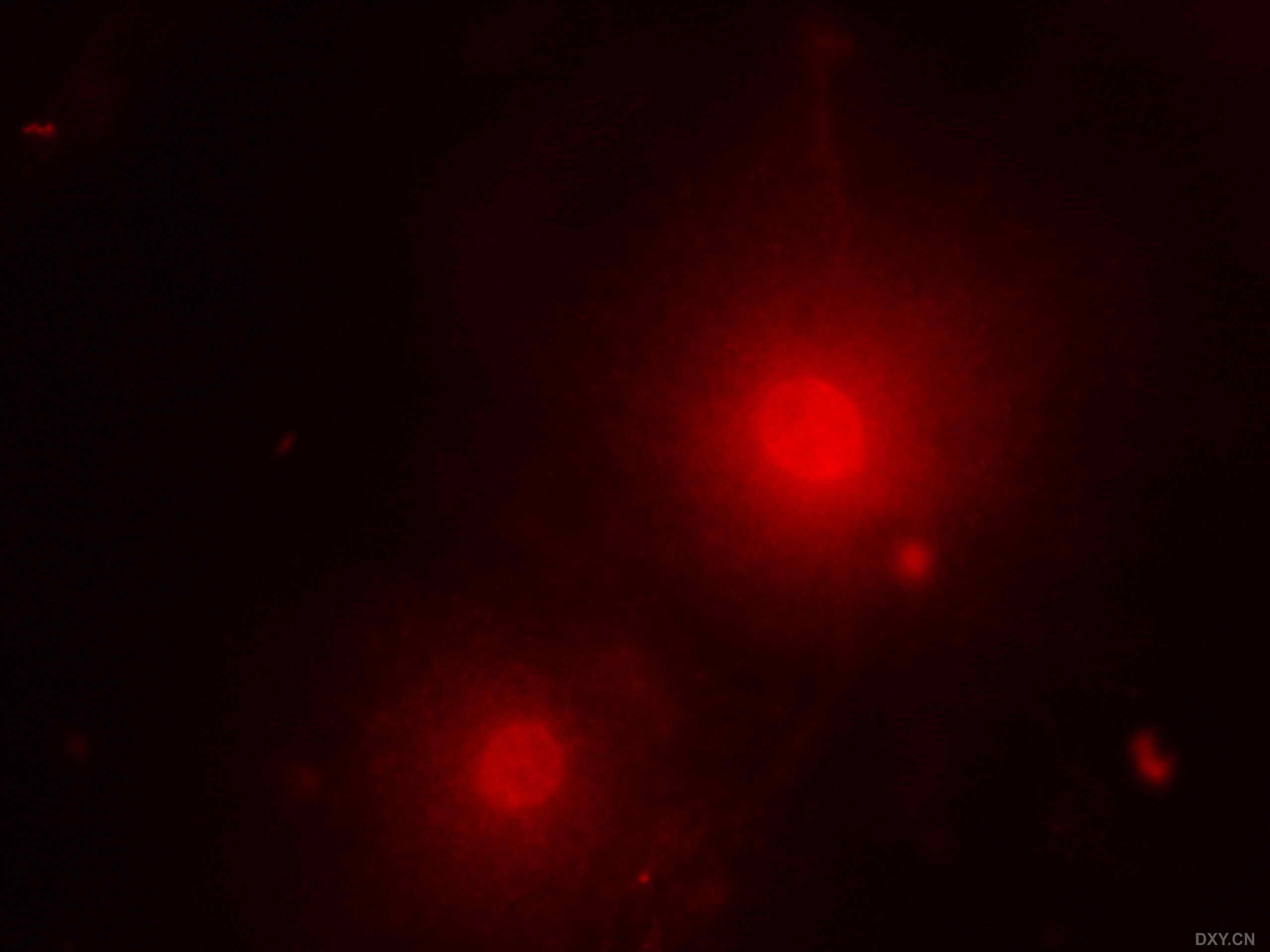 图片附件: 73905940.jpg (2011-12-6 16:57, 16.6 KB) / 该附件被下载次数 4
图片附件: 73905940.jpg (2011-12-6 16:57, 16.6 KB) / 该附件被下载次数 4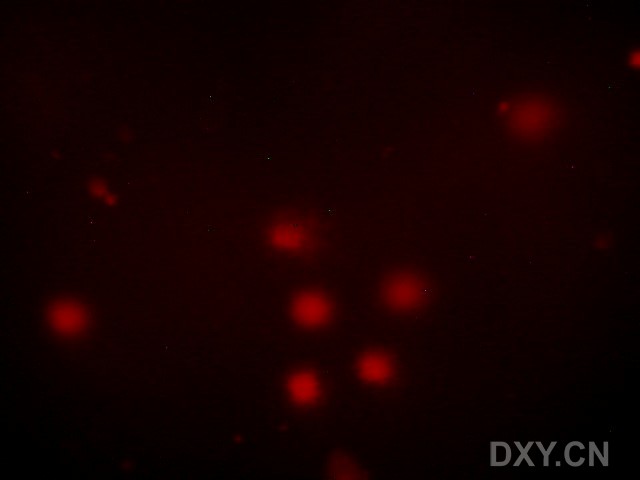 图片附件: 40763344.jpg (2011-12-6 16:57, 16.17 KB) / 该附件被下载次数 6
图片附件: 40763344.jpg (2011-12-6 16:57, 16.17 KB) / 该附件被下载次数 6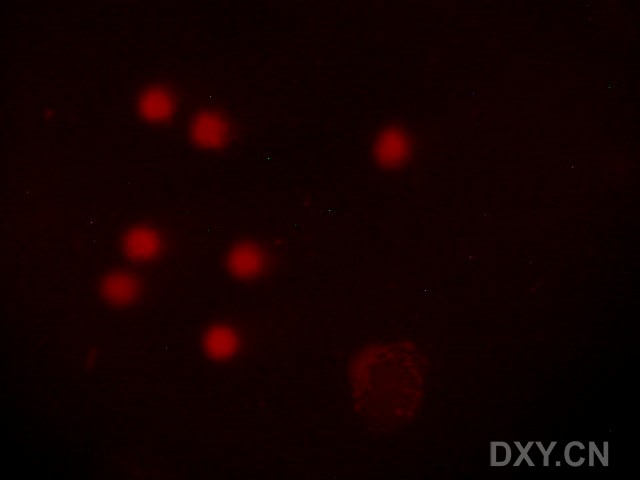 图片附件: 48491100.jpg (2011-12-6 16:57, 20.89 KB) / 该附件被下载次数 3
图片附件: 48491100.jpg (2011-12-6 16:57, 20.89 KB) / 该附件被下载次数 3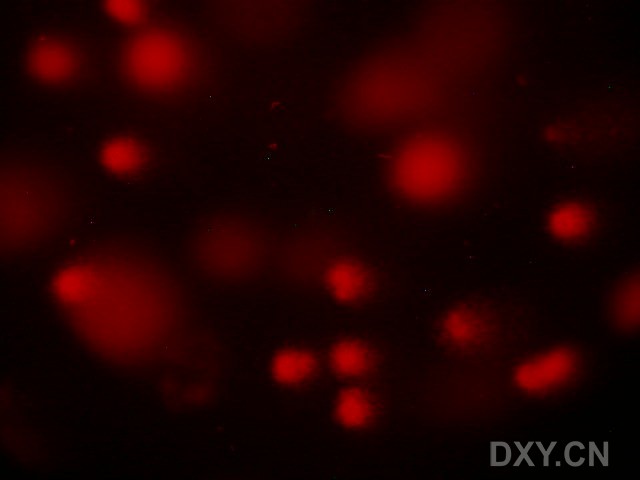 图片附件: 21687122.jpg (2011-12-6 17:07, 26.58 KB) / 该附件被下载次数 2
图片附件: 21687122.jpg (2011-12-6 17:07, 26.58 KB) / 该附件被下载次数 2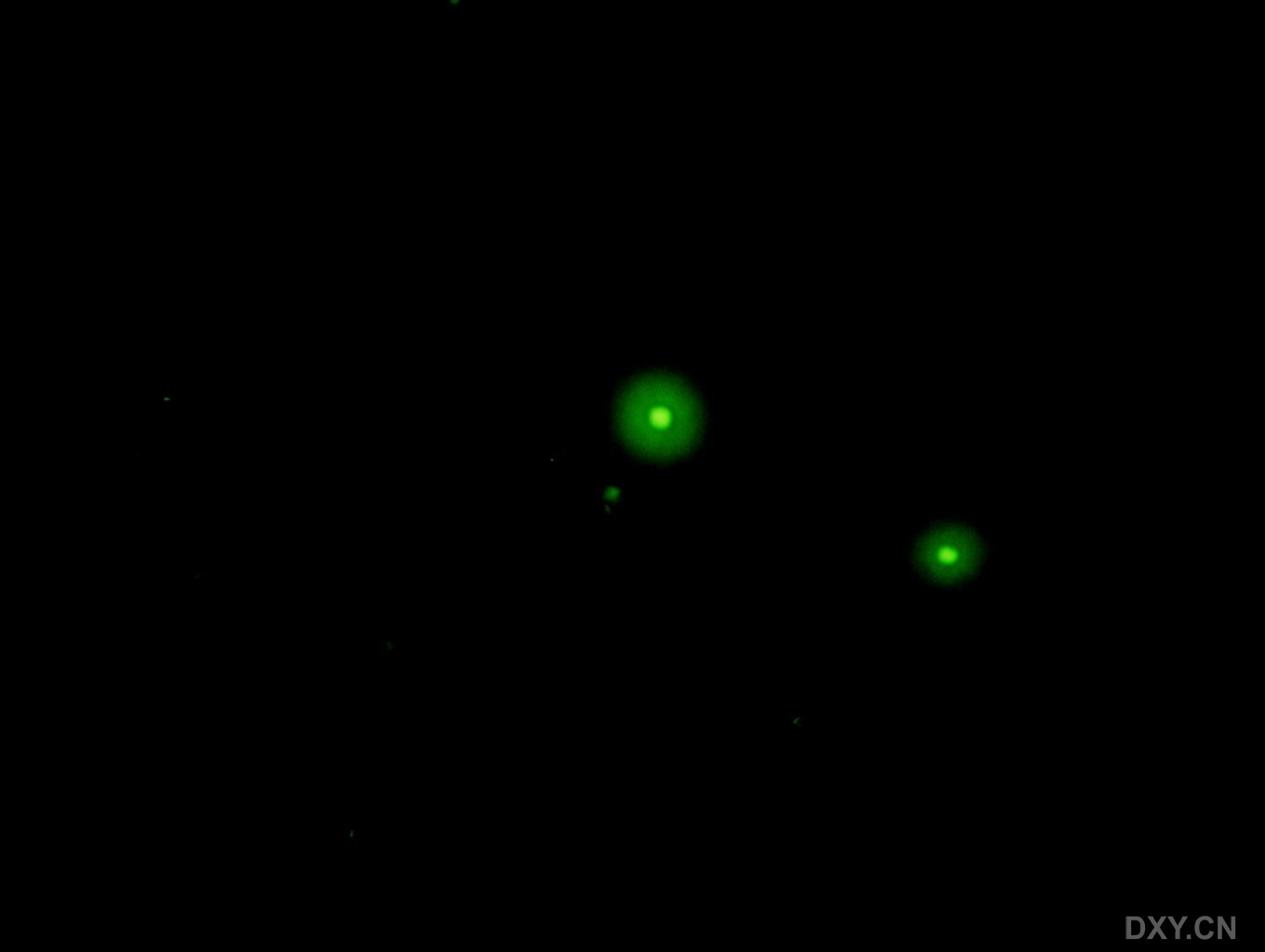 图片附件: 16704945.jpg (2011-12-6 17:07, 27.81 KB) / 该附件被下载次数 1
图片附件: 16704945.jpg (2011-12-6 17:07, 27.81 KB) / 该附件被下载次数 1 图片附件: 88051376.snap.jpg (2011-12-6 17:07, 12.83 KB) / 该附件被下载次数 5
图片附件: 88051376.snap.jpg (2011-12-6 17:07, 12.83 KB) / 该附件被下载次数 5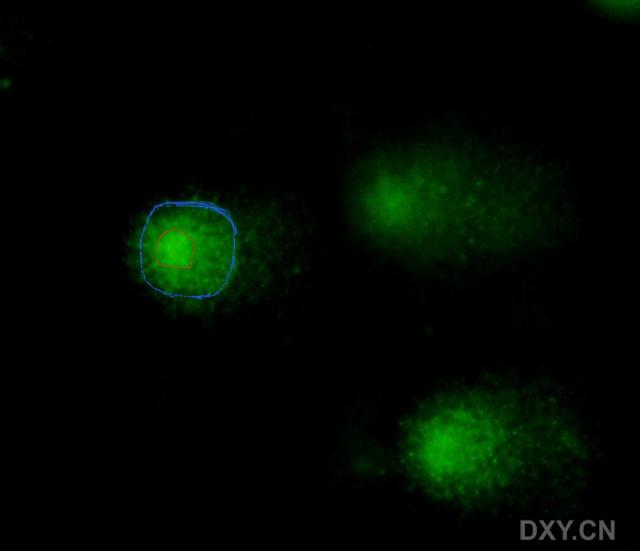 图片附件: 17352706.snap.jpg (2011-12-6 17:08, 12.45 KB) / 该附件被下载次数 4
图片附件: 17352706.snap.jpg (2011-12-6 17:08, 12.45 KB) / 该附件被下载次数 4 图片附件: 71920380.jpg (2011-12-6 17:18, 12.9 KB) / 该附件被下载次数 3
图片附件: 71920380.jpg (2011-12-6 17:18, 12.9 KB) / 该附件被下载次数 3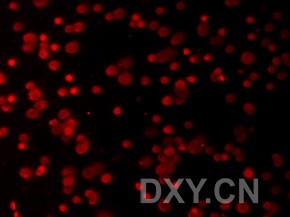 图片附件: 45523149.snap.jpg (2011-12-6 17:29, 8.45 KB) / 该附件被下载次数 5
图片附件: 45523149.snap.jpg (2011-12-6 17:29, 8.45 KB) / 该附件被下载次数 5 图片附件: 35379681.jpg (2011-12-6 17:36, 31.95 KB) / 该附件被下载次数 7
图片附件: 35379681.jpg (2011-12-6 17:36, 31.95 KB) / 该附件被下载次数 7 图片附件: 10671681.snap.jpg (2011-12-7 10:29, 11.06 KB) / 该附件被下载次数 7
图片附件: 10671681.snap.jpg (2011-12-7 10:29, 11.06 KB) / 该附件被下载次数 7 图片附件: 56861829.snap.jpg (2011-12-7 10:30, 9.86 KB) / 该附件被下载次数 3
图片附件: 56861829.snap.jpg (2011-12-7 10:30, 9.86 KB) / 该附件被下载次数 3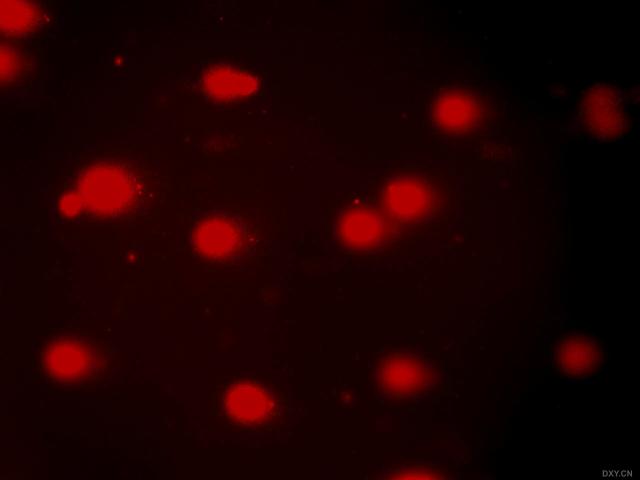 图片附件: 90745648.snap.jpg (2011-12-7 10:31, 26.86 KB) / 该附件被下载次数 3
图片附件: 90745648.snap.jpg (2011-12-7 10:31, 26.86 KB) / 该附件被下载次数 3 图片附件: 70654918.jpg (2011-12-7 10:37, 18.23 KB) / 该附件被下载次数 9
图片附件: 70654918.jpg (2011-12-7 10:37, 18.23 KB) / 该附件被下载次数 9 图片附件: 28285955.snap.jpg (2011-12-7 10:51, 12.57 KB) / 该附件被下载次数 4
图片附件: 28285955.snap.jpg (2011-12-7 10:51, 12.57 KB) / 该附件被下载次数 4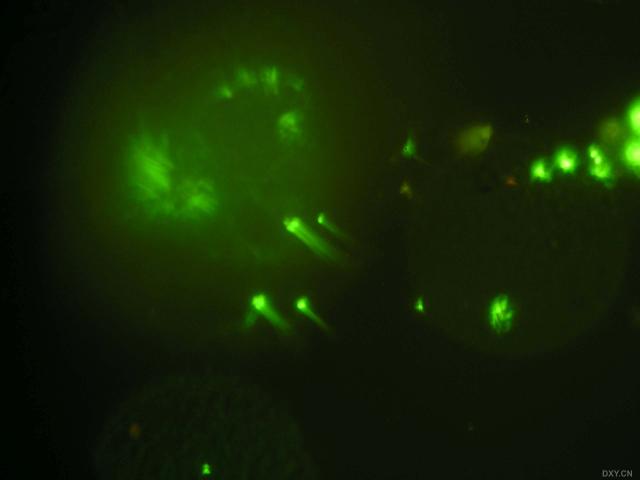 图片附件: 31718979.snap.jpg (2011-12-7 10:52, 15.04 KB) / 该附件被下载次数 6
图片附件: 31718979.snap.jpg (2011-12-7 10:52, 15.04 KB) / 该附件被下载次数 6 图片附件: 18335811.jpg (2011-12-7 11:00, 16.49 KB) / 该附件被下载次数 2
图片附件: 18335811.jpg (2011-12-7 11:00, 16.49 KB) / 该附件被下载次数 2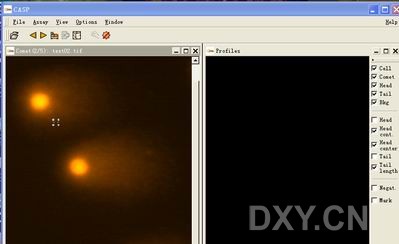 图片附件: 75583300.jpg (2011-12-7 11:05, 97.26 KB) / 该附件被下载次数 4
图片附件: 75583300.jpg (2011-12-7 11:05, 97.26 KB) / 该附件被下载次数 4 图片附件: 80041967.jpg (2011-12-7 11:05, 106.91 KB) / 该附件被下载次数 2
图片附件: 80041967.jpg (2011-12-7 11:05, 106.91 KB) / 该附件被下载次数 2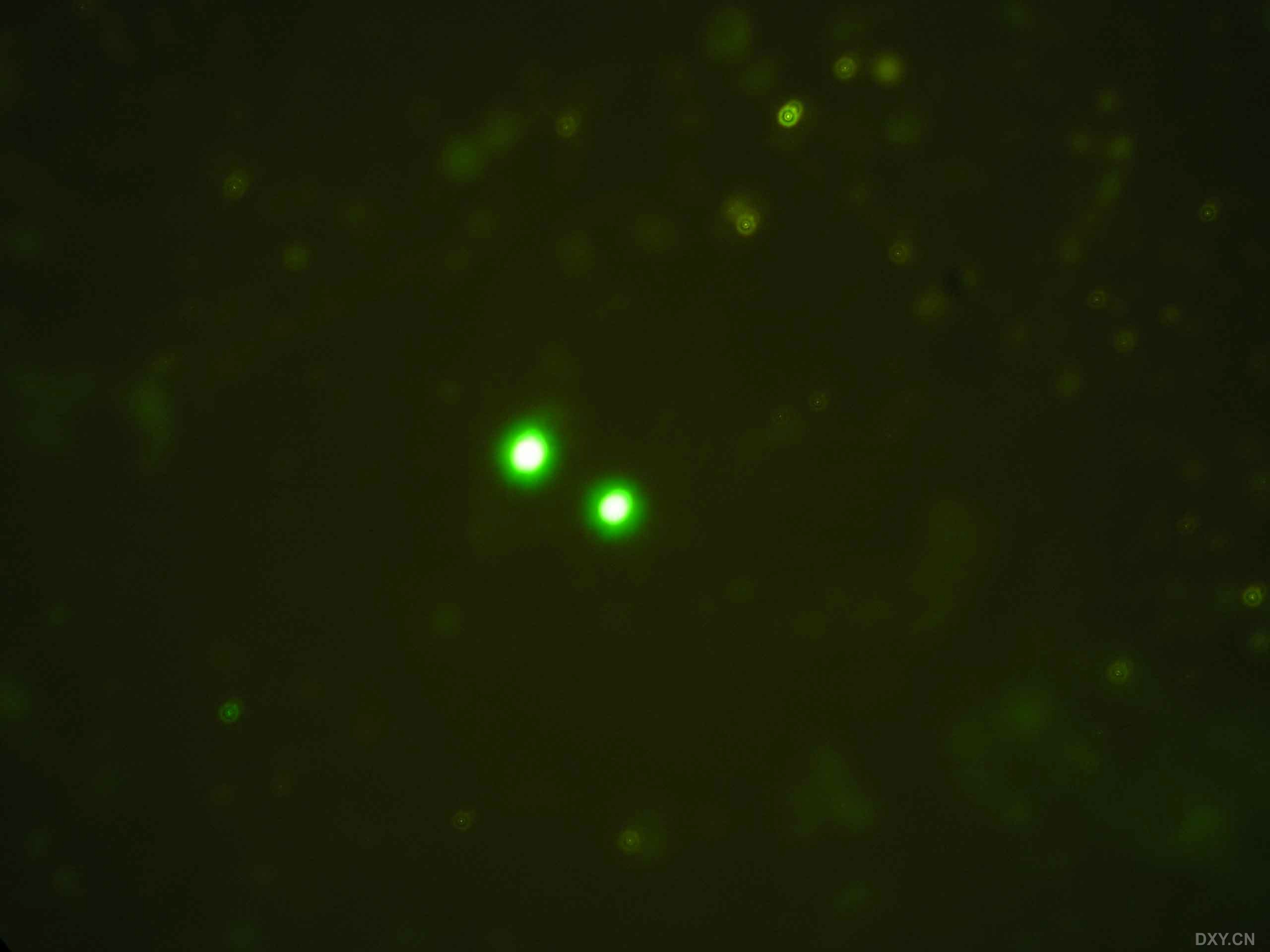 图片附件: 75390684.snap.jpg (2011-12-7 11:09, 10.6 KB) / 该附件被下载次数 4
图片附件: 75390684.snap.jpg (2011-12-7 11:09, 10.6 KB) / 该附件被下载次数 4 图片附件: 16179891.jpg (2011-12-7 11:11, 15.2 KB) / 该附件被下载次数 3
图片附件: 16179891.jpg (2011-12-7 11:11, 15.2 KB) / 该附件被下载次数 3 图片附件: 78073094.jpg (2011-12-7 11:12, 14.97 KB) / 该附件被下载次数 6
图片附件: 78073094.jpg (2011-12-7 11:12, 14.97 KB) / 该附件被下载次数 6 图片附件: 63284107.jpg (2011-12-7 11:13, 74 KB) / 该附件被下载次数 3
图片附件: 63284107.jpg (2011-12-7 11:13, 74 KB) / 该附件被下载次数 3 图片附件: 96463957.jpg (2011-12-7 11:14, 80.61 KB) / 该附件被下载次数 3
图片附件: 96463957.jpg (2011-12-7 11:14, 80.61 KB) / 该附件被下载次数 3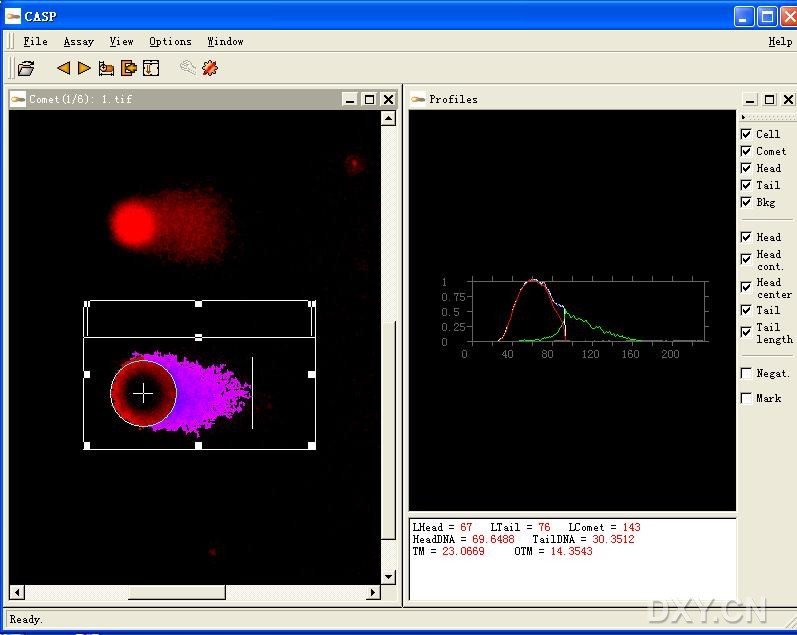 图片附件: 39137653.snap.jpg (2011-12-7 11:17, 6.61 KB) / 该附件被下载次数 6
图片附件: 39137653.snap.jpg (2011-12-7 11:17, 6.61 KB) / 该附件被下载次数 6 图片附件: 12203077.jpg (2011-12-7 14:31, 8.97 KB) / 该附件被下载次数 3
图片附件: 12203077.jpg (2011-12-7 14:31, 8.97 KB) / 该附件被下载次数 3 图片附件: 56397818.jpg (2011-12-7 14:46, 6.28 KB) / 该附件被下载次数 4
图片附件: 56397818.jpg (2011-12-7 14:46, 6.28 KB) / 该附件被下载次数 4 图片附件: 25323030.jpg (2011-12-7 14:58, 60.32 KB) / 该附件被下载次数 5
图片附件: 25323030.jpg (2011-12-7 14:58, 60.32 KB) / 该附件被下载次数 5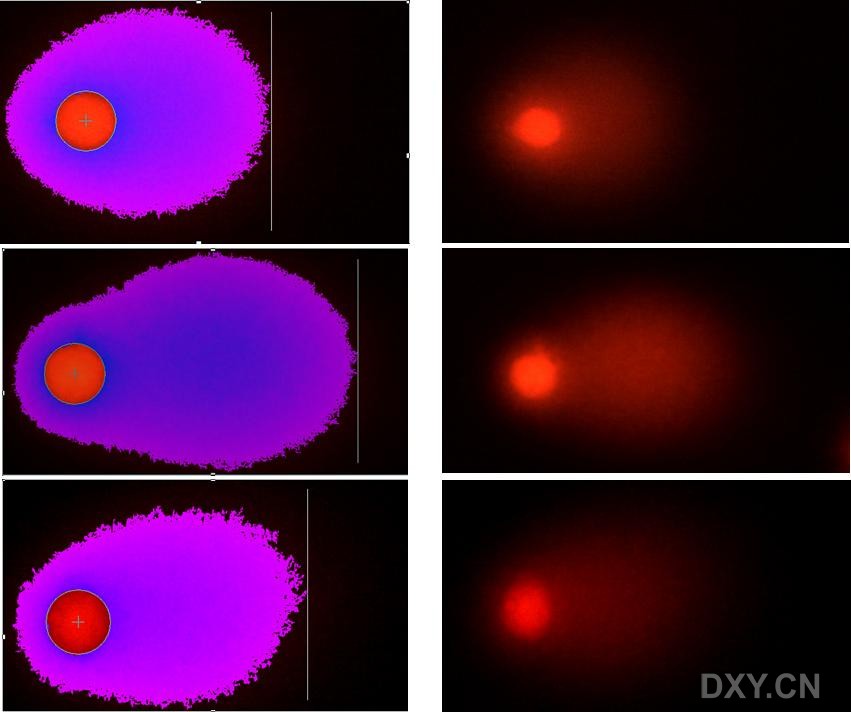 图片附件: 39372644.jpg (2011-12-7 15:08, 10.14 KB) / 该附件被下载次数 2
图片附件: 39372644.jpg (2011-12-7 15:08, 10.14 KB) / 该附件被下载次数 2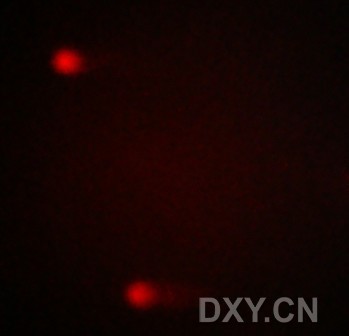 图片附件: 36225687.jpg (2011-12-7 15:08, 16.8 KB) / 该附件被下载次数 3
图片附件: 36225687.jpg (2011-12-7 15:08, 16.8 KB) / 该附件被下载次数 3 图片附件: 27154870.jpg (2011-12-7 15:24, 12.46 KB) / 该附件被下载次数 6
图片附件: 27154870.jpg (2011-12-7 15:24, 12.46 KB) / 该附件被下载次数 6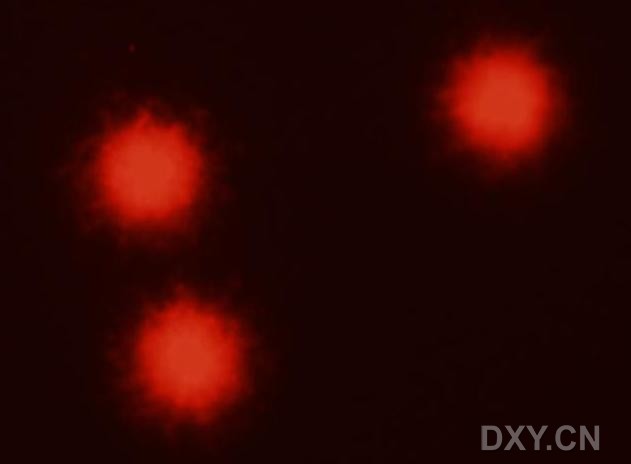 图片附件: 33633981.jpg (2011-12-7 15:24, 12.11 KB) / 该附件被下载次数 4
图片附件: 33633981.jpg (2011-12-7 15:24, 12.11 KB) / 该附件被下载次数 4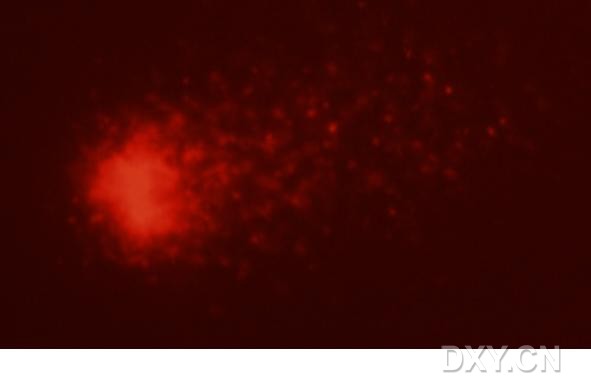 图片附件: 20965241.snap.jpg (2011-12-7 15:27, 6.13 KB) / 该附件被下载次数 1
图片附件: 20965241.snap.jpg (2011-12-7 15:27, 6.13 KB) / 该附件被下载次数 1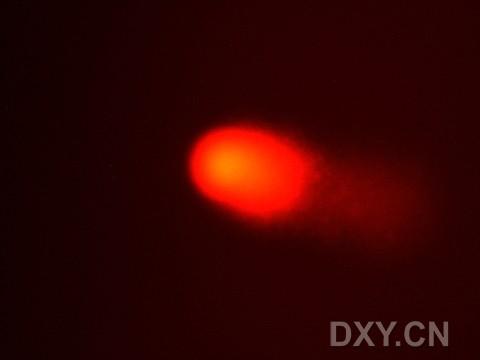 图片附件: 78459922.snap.jpg (2011-12-7 15:27, 5.71 KB) / 该附件被下载次数 3
图片附件: 78459922.snap.jpg (2011-12-7 15:27, 5.71 KB) / 该附件被下载次数 3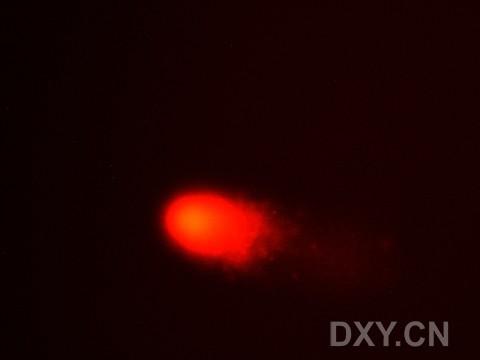 图片附件: 86892796.snap.jpg (2011-12-7 15:27, 6.08 KB) / 该附件被下载次数 3
图片附件: 86892796.snap.jpg (2011-12-7 15:27, 6.08 KB) / 该附件被下载次数 3 图片附件: 30973870.snap.jpg (2011-12-7 15:28, 49.32 KB) / 该附件被下载次数 4
图片附件: 30973870.snap.jpg (2011-12-7 15:28, 49.32 KB) / 该附件被下载次数 4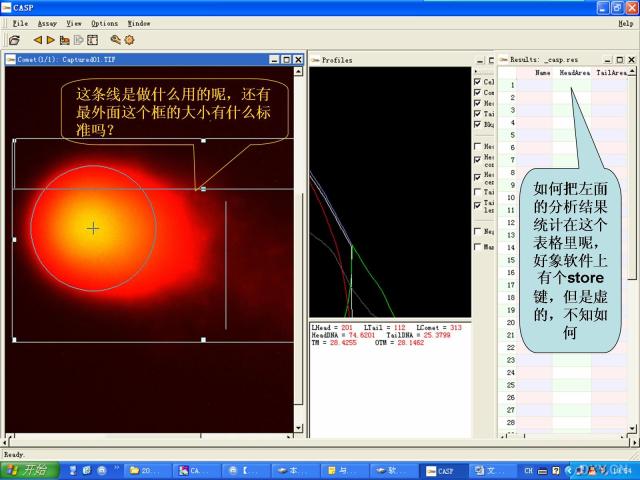 图片附件: 83054601.snap.jpg (2011-12-7 15:50, 12.26 KB) / 该附件被下载次数 0
图片附件: 83054601.snap.jpg (2011-12-7 15:50, 12.26 KB) / 该附件被下载次数 0 图片附件: 22631488.snap.jpg (2011-12-7 16:05, 36.62 KB) / 该附件被下载次数 0
图片附件: 22631488.snap.jpg (2011-12-7 16:05, 36.62 KB) / 该附件被下载次数 0 图片附件: 72090968.snap.jpg (2011-12-7 16:05, 16.39 KB) / 该附件被下载次数 4
图片附件: 72090968.snap.jpg (2011-12-7 16:05, 16.39 KB) / 该附件被下载次数 4 图片附件: 34791418.snap.jpg (2011-12-7 16:05, 15.53 KB) / 该附件被下载次数 4
图片附件: 34791418.snap.jpg (2011-12-7 16:05, 15.53 KB) / 该附件被下载次数 4 图片附件: 54481294.snap.jpg (2011-12-7 16:06, 6.37 KB) / 该附件被下载次数 3
图片附件: 54481294.snap.jpg (2011-12-7 16:06, 6.37 KB) / 该附件被下载次数 3 图片附件: 20976622.snap.jpg (2011-12-7 16:06, 6.2 KB) / 该附件被下载次数 6
图片附件: 20976622.snap.jpg (2011-12-7 16:06, 6.2 KB) / 该附件被下载次数 6 图片附件: 32048908.snap.jpg (2011-12-7 16:06, 5.93 KB) / 该附件被下载次数 2
图片附件: 32048908.snap.jpg (2011-12-7 16:06, 5.93 KB) / 该附件被下载次数 2 图片附件: 83591900.snap.jpg (2011-12-7 16:11, 6.28 KB) / 该附件被下载次数 3
图片附件: 83591900.snap.jpg (2011-12-7 16:11, 6.28 KB) / 该附件被下载次数 3 图片附件: 74281333.snap.jpg (2011-12-7 16:11, 6.29 KB) / 该附件被下载次数 6
图片附件: 74281333.snap.jpg (2011-12-7 16:11, 6.29 KB) / 该附件被下载次数 6 图片附件: 46771126.snap.jpg (2011-12-7 16:17, 20.98 KB) / 该附件被下载次数 3
图片附件: 46771126.snap.jpg (2011-12-7 16:17, 20.98 KB) / 该附件被下载次数 3 图片附件: 35923928.snap.jpg (2011-12-7 16:17, 34.12 KB) / 该附件被下载次数 3
图片附件: 35923928.snap.jpg (2011-12-7 16:17, 34.12 KB) / 该附件被下载次数 3 图片附件: 40110449.jpg (2011-12-7 16:45, 51.77 KB) / 该附件被下载次数 3
图片附件: 40110449.jpg (2011-12-7 16:45, 51.77 KB) / 该附件被下载次数 3 图片附件: 30372705.snap.jpg (2011-12-7 16:45, 10.61 KB) / 该附件被下载次数 2
图片附件: 30372705.snap.jpg (2011-12-7 16:45, 10.61 KB) / 该附件被下载次数 2 图片附件: 64283651.snap.jpg (2011-12-7 16:47, 31.21 KB) / 该附件被下载次数 5
图片附件: 64283651.snap.jpg (2011-12-7 16:47, 31.21 KB) / 该附件被下载次数 5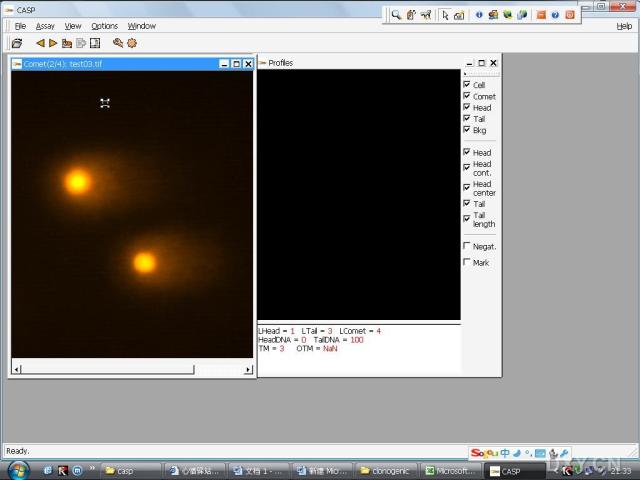 图片附件: 11966463.jpg (2011-12-7 17:00, 39.6 KB) / 该附件被下载次数 1
图片附件: 11966463.jpg (2011-12-7 17:00, 39.6 KB) / 该附件被下载次数 1 图片附件: 96520868.jpg (2011-12-7 17:01, 21.57 KB) / 该附件被下载次数 2
图片附件: 96520868.jpg (2011-12-7 17:01, 21.57 KB) / 该附件被下载次数 2 图片附件: 41591040.snap.jpg (2011-12-7 17:04, 10.74 KB) / 该附件被下载次数 3
图片附件: 41591040.snap.jpg (2011-12-7 17:04, 10.74 KB) / 该附件被下载次数 3 图片附件: 92419234.snap.jpg (2011-12-7 17:11, 11.59 KB) / 该附件被下载次数 0
图片附件: 92419234.snap.jpg (2011-12-7 17:11, 11.59 KB) / 该附件被下载次数 0 图片附件: 42978153.snap.jpg (2011-12-7 17:11, 10.76 KB) / 该附件被下载次数 2
图片附件: 42978153.snap.jpg (2011-12-7 17:11, 10.76 KB) / 该附件被下载次数 2 图片附件: 27313077.snap.jpg (2011-12-7 17:11, 9.32 KB) / 该附件被下载次数 2
图片附件: 27313077.snap.jpg (2011-12-7 17:11, 9.32 KB) / 该附件被下载次数 2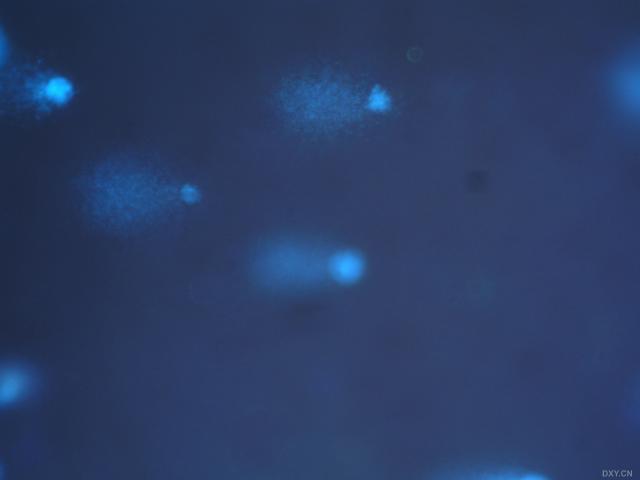 图片附件: 28062862.snap.jpg (2011-12-7 17:12, 6.51 KB) / 该附件被下载次数 1
图片附件: 28062862.snap.jpg (2011-12-7 17:12, 6.51 KB) / 该附件被下载次数 1 图片附件: 32099555.snap.jpg (2011-12-7 17:15, 9 KB) / 该附件被下载次数 5
图片附件: 32099555.snap.jpg (2011-12-7 17:15, 9 KB) / 该附件被下载次数 5 图片附件: 76080706.snap.jpg (2011-12-7 17:15, 9.01 KB) / 该附件被下载次数 2
图片附件: 76080706.snap.jpg (2011-12-7 17:15, 9.01 KB) / 该附件被下载次数 2 图片附件: 21155728.snap.jpg (2011-12-7 17:16, 9.84 KB) / 该附件被下载次数 2
图片附件: 21155728.snap.jpg (2011-12-7 17:16, 9.84 KB) / 该附件被下载次数 2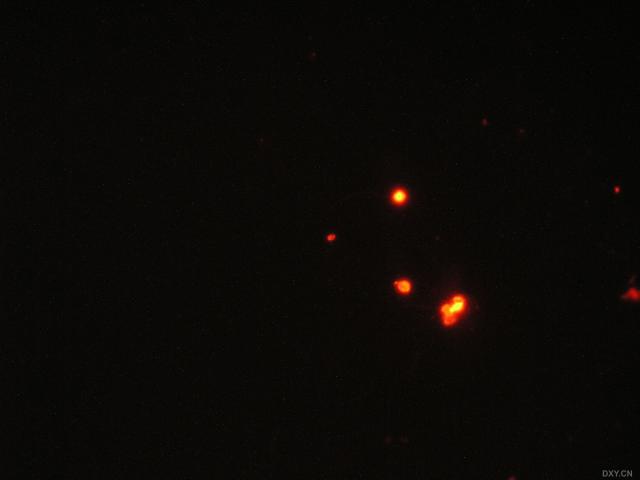 图片附件: 55464899.snap.jpg (2011-12-7 17:17, 9.52 KB) / 该附件被下载次数 4
图片附件: 55464899.snap.jpg (2011-12-7 17:17, 9.52 KB) / 该附件被下载次数 4 图片附件: 17749952.snap.jpg (2011-12-7 17:21, 18.6 KB) / 该附件被下载次数 3
图片附件: 17749952.snap.jpg (2011-12-7 17:21, 18.6 KB) / 该附件被下载次数 3 图片附件: 72149538.snap.jpg (2011-12-7 17:21, 9.77 KB) / 该附件被下载次数 0
图片附件: 72149538.snap.jpg (2011-12-7 17:21, 9.77 KB) / 该附件被下载次数 0 图片附件: 49877858.snap.jpg (2011-12-7 17:22, 24.61 KB) / 该附件被下载次数 3
图片附件: 49877858.snap.jpg (2011-12-7 17:22, 24.61 KB) / 该附件被下载次数 3 图片附件: 60312376.png (2011-12-7 17:24, 52.31 KB) / 该附件被下载次数 3
图片附件: 60312376.png (2011-12-7 17:24, 52.31 KB) / 该附件被下载次数 3 图片附件: 35711335.snap.jpg (2011-12-7 17:24, 9.22 KB) / 该附件被下载次数 3
图片附件: 35711335.snap.jpg (2011-12-7 17:24, 9.22 KB) / 该附件被下载次数 3 图片附件: 46536612.snap.jpg (2011-12-7 17:25, 3.85 KB) / 该附件被下载次数 2
图片附件: 46536612.snap.jpg (2011-12-7 17:25, 3.85 KB) / 该附件被下载次数 2 图片附件: 67847225.snap.jpg (2011-12-7 17:25, 16.91 KB) / 该附件被下载次数 5
图片附件: 67847225.snap.jpg (2011-12-7 17:25, 16.91 KB) / 该附件被下载次数 5 图片附件: 66590823.snap.jpg (2011-12-7 17:27, 23.17 KB) / 该附件被下载次数 3
图片附件: 66590823.snap.jpg (2011-12-7 17:27, 23.17 KB) / 该附件被下载次数 3 图片附件: 31040049.snap.jpg (2011-12-8 08:32, 9.65 KB) / 该附件被下载次数 3
图片附件: 31040049.snap.jpg (2011-12-8 08:32, 9.65 KB) / 该附件被下载次数 3 图片附件: 90158792.snap.jpg (2011-12-8 10:11, 11.8 KB) / 该附件被下载次数 5
图片附件: 90158792.snap.jpg (2011-12-8 10:11, 11.8 KB) / 该附件被下载次数 5 图片附件: 22753855.snap.jpg (2011-12-8 10:33, 7.93 KB) / 该附件被下载次数 4
图片附件: 22753855.snap.jpg (2011-12-8 10:33, 7.93 KB) / 该附件被下载次数 4 图片附件: 48143049.snap.jpg (2011-12-8 10:34, 10.08 KB) / 该附件被下载次数 5
图片附件: 48143049.snap.jpg (2011-12-8 10:34, 10.08 KB) / 该附件被下载次数 5 图片附件: 43198853.jpg (2011-12-8 10:35, 20.13 KB) / 该附件被下载次数 7
图片附件: 43198853.jpg (2011-12-8 10:35, 20.13 KB) / 该附件被下载次数 7 图片附件: 33890923.snap.jpg (2011-12-8 10:36, 5.75 KB) / 该附件被下载次数 8
图片附件: 33890923.snap.jpg (2011-12-8 10:36, 5.75 KB) / 该附件被下载次数 8 图片附件: 51316885.snap.jpg (2011-12-8 10:36, 33.58 KB) / 该附件被下载次数 5
图片附件: 51316885.snap.jpg (2011-12-8 10:36, 33.58 KB) / 该附件被下载次数 5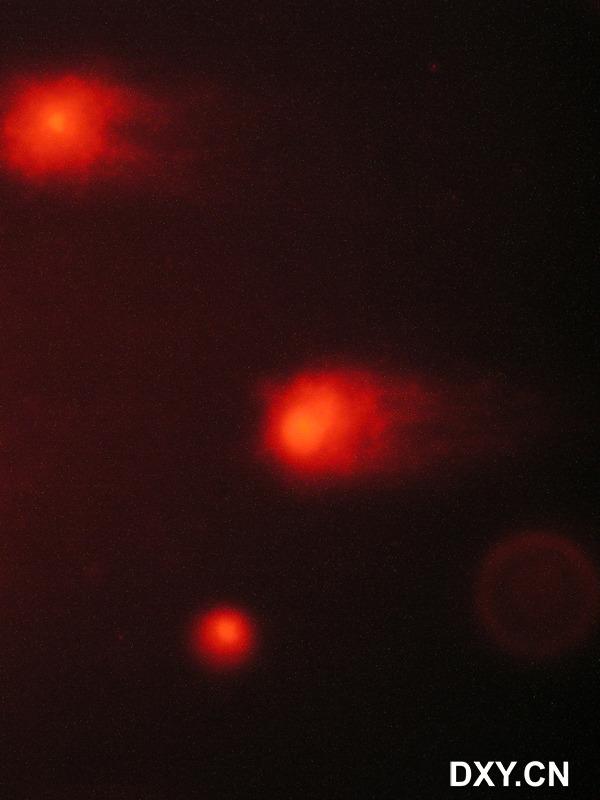 图片附件: 16124522.snap.jpg (2011-12-8 10:37, 27.35 KB) / 该附件被下载次数 8
图片附件: 16124522.snap.jpg (2011-12-8 10:37, 27.35 KB) / 该附件被下载次数 8 图片附件: 91699515.jpg (2011-12-8 11:14, 11.88 KB) / 该附件被下载次数 8
图片附件: 91699515.jpg (2011-12-8 11:14, 11.88 KB) / 该附件被下载次数 8 图片附件: 94031090.jpg (2011-12-8 11:19, 4.14 KB) / 该附件被下载次数 5
图片附件: 94031090.jpg (2011-12-8 11:19, 4.14 KB) / 该附件被下载次数 5 图片附件: 79344209.snap.jpg (2011-12-8 11:19, 78.94 KB) / 该附件被下载次数 4
图片附件: 79344209.snap.jpg (2011-12-8 11:19, 78.94 KB) / 该附件被下载次数 4 图片附件: 72007676.snap.jpg (2011-12-8 11:19, 80.57 KB) / 该附件被下载次数 4
图片附件: 72007676.snap.jpg (2011-12-8 11:19, 80.57 KB) / 该附件被下载次数 4 图片附件: 57311372.snap.jpg (2011-12-8 14:53, 219.23 KB) / 该附件被下载次数 5
图片附件: 57311372.snap.jpg (2011-12-8 14:53, 219.23 KB) / 该附件被下载次数 5 图片附件: 30919424.snap.jpg (2011-12-8 14:53, 241.84 KB) / 该附件被下载次数 4
图片附件: 30919424.snap.jpg (2011-12-8 14:53, 241.84 KB) / 该附件被下载次数 4 图片附件: 32052303.snap.jpg (2011-12-8 14:54, 202.71 KB) / 该附件被下载次数 4
图片附件: 32052303.snap.jpg (2011-12-8 14:54, 202.71 KB) / 该附件被下载次数 4 图片附件: 96095669.snap.jpg (2011-12-8 14:54, 187.65 KB) / 该附件被下载次数 2
图片附件: 96095669.snap.jpg (2011-12-8 14:54, 187.65 KB) / 该附件被下载次数 2 图片附件: 50298004.snap.jpg (2011-12-8 14:54, 200.56 KB) / 该附件被下载次数 5
图片附件: 50298004.snap.jpg (2011-12-8 14:54, 200.56 KB) / 该附件被下载次数 5 图片附件: 57454251.jpg (2011-12-8 14:57, 11.58 KB) / 该附件被下载次数 4
图片附件: 57454251.jpg (2011-12-8 14:57, 11.58 KB) / 该附件被下载次数 4 图片附件: 83293896.jpg (2011-12-8 14:57, 10.34 KB) / 该附件被下载次数 4
图片附件: 83293896.jpg (2011-12-8 14:57, 10.34 KB) / 该附件被下载次数 4 图片附件: 13827680.snap.jpg (2011-12-8 15:04, 90.68 KB) / 该附件被下载次数 3
图片附件: 13827680.snap.jpg (2011-12-8 15:04, 90.68 KB) / 该附件被下载次数 3 图片附件: 68358677.snap.jpg (2011-12-8 15:10, 102.91 KB) / 该附件被下载次数 3
图片附件: 68358677.snap.jpg (2011-12-8 15:10, 102.91 KB) / 该附件被下载次数 3 图片附件: 92688338.snap.jpg (2011-12-8 15:11, 94.3 KB) / 该附件被下载次数 5
图片附件: 92688338.snap.jpg (2011-12-8 15:11, 94.3 KB) / 该附件被下载次数 5 图片附件: 17079533.snap.jpg (2011-12-8 15:18, 5.58 KB) / 该附件被下载次数 2
图片附件: 17079533.snap.jpg (2011-12-8 15:18, 5.58 KB) / 该附件被下载次数 2 图片附件: 72210924.snap.jpg (2011-12-8 15:18, 6.49 KB) / 该附件被下载次数 2
图片附件: 72210924.snap.jpg (2011-12-8 15:18, 6.49 KB) / 该附件被下载次数 2 图片附件: 63739124.snap.jpg (2011-12-8 15:18, 24.02 KB) / 该附件被下载次数 7
图片附件: 63739124.snap.jpg (2011-12-8 15:18, 24.02 KB) / 该附件被下载次数 7 图片附件: 96443605.snap.jpg (2011-12-8 15:21, 30.77 KB) / 该附件被下载次数 3
图片附件: 96443605.snap.jpg (2011-12-8 15:21, 30.77 KB) / 该附件被下载次数 3 图片附件: 78293554.snap.jpg (2011-12-8 15:21, 23.57 KB) / 该附件被下载次数 5
图片附件: 78293554.snap.jpg (2011-12-8 15:21, 23.57 KB) / 该附件被下载次数 5
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |