 图片附件: 33189372.jpg (2012-1-19 14:39, 52.33 KB) / 该附件被下载次数 25
图片附件: 33189372.jpg (2012-1-19 14:39, 52.33 KB) / 该附件被下载次数 25 图片附件: 51570500.snap.jpg (2012-1-19 14:39, 50.02 KB) / 该附件被下载次数 22
图片附件: 51570500.snap.jpg (2012-1-19 14:39, 50.02 KB) / 该附件被下载次数 22 图片附件: 90064363.snap.jpg (2012-1-19 14:40, 45.39 KB) / 该附件被下载次数 28
图片附件: 90064363.snap.jpg (2012-1-19 14:40, 45.39 KB) / 该附件被下载次数 28 图片附件: 22478679.snap.jpg (2012-1-19 14:40, 53.87 KB) / 该附件被下载次数 24
图片附件: 22478679.snap.jpg (2012-1-19 14:40, 53.87 KB) / 该附件被下载次数 24 图片附件: 51085--embed.jpg (2012-1-19 14:51, 202.61 KB) / 该附件被下载次数 11
图片附件: 51085--embed.jpg (2012-1-19 14:51, 202.61 KB) / 该附件被下载次数 11 图片附件: 88423-2-embed.jpg (2012-1-19 16:11, 13.56 KB) / 该附件被下载次数 9
图片附件: 88423-2-embed.jpg (2012-1-19 16:11, 13.56 KB) / 该附件被下载次数 9 图片附件: 132090-ResizeofResizeofMSCCYTOMETRYpart-embed (1).jpg (2012-1-21 09:33, 30.61 KB) / 该附件被下载次数 6
图片附件: 132090-ResizeofResizeofMSCCYTOMETRYpart-embed (1).jpg (2012-1-21 09:33, 30.61 KB) / 该附件被下载次数 6 图片附件: 150239-endo-2-embed.gif (2012-1-21 10:37, 19.36 KB) / 该附件被下载次数 8
图片附件: 150239-endo-2-embed.gif (2012-1-21 10:37, 19.36 KB) / 该附件被下载次数 8 图片附件: 80977951.jpg (2012-1-21 11:20, 47.05 KB) / 该附件被下载次数 3
图片附件: 80977951.jpg (2012-1-21 11:20, 47.05 KB) / 该附件被下载次数 3 图片附件: 30457779.snap.jpg (2012-1-21 11:25, 65.92 KB) / 该附件被下载次数 7
图片附件: 30457779.snap.jpg (2012-1-21 11:25, 65.92 KB) / 该附件被下载次数 7 图片附件: 46868233.jpg (2012-1-21 11:35, 43.71 KB) / 该附件被下载次数 3
图片附件: 46868233.jpg (2012-1-21 11:35, 43.71 KB) / 该附件被下载次数 3 图片附件: 84503630.jpg (2012-1-21 12:53, 19.75 KB) / 该附件被下载次数 6
图片附件: 84503630.jpg (2012-1-21 12:53, 19.75 KB) / 该附件被下载次数 6 图片附件: 87557544_snap.jpg (2012-1-28 09:09, 28.19 KB) / 该附件被下载次数 4
图片附件: 87557544_snap.jpg (2012-1-28 09:09, 28.19 KB) / 该附件被下载次数 4 图片附件: 38600332.snap.jpg (2012-1-28 09:12, 29.75 KB) / 该附件被下载次数 6
图片附件: 38600332.snap.jpg (2012-1-28 09:12, 29.75 KB) / 该附件被下载次数 6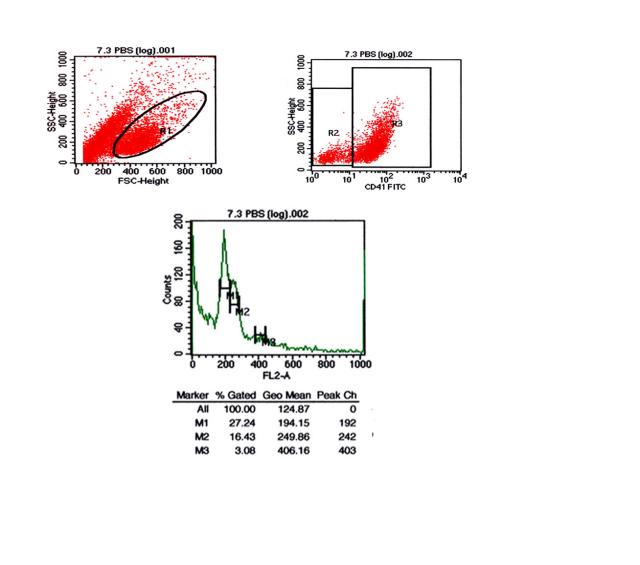 图片附件: 22042763.snap.jpg (2012-1-28 09:12, 26.42 KB) / 该附件被下载次数 6
图片附件: 22042763.snap.jpg (2012-1-28 09:12, 26.42 KB) / 该附件被下载次数 6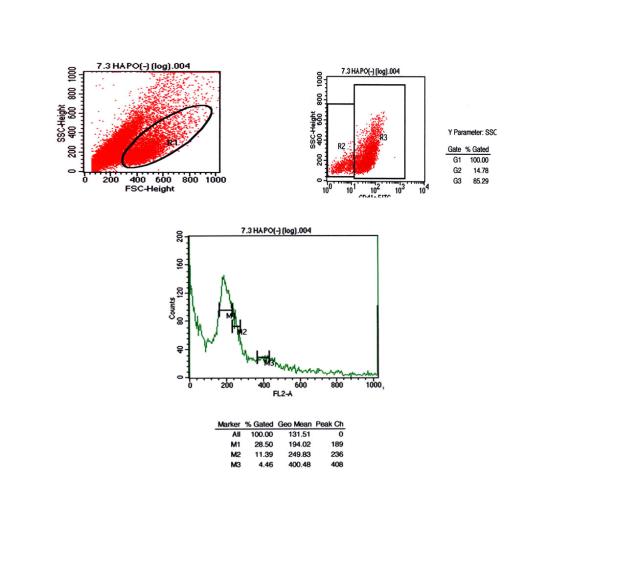 图片附件: 91091591.snap.jpg (2012-1-28 09:58, 34.98 KB) / 该附件被下载次数 6
图片附件: 91091591.snap.jpg (2012-1-28 09:58, 34.98 KB) / 该附件被下载次数 6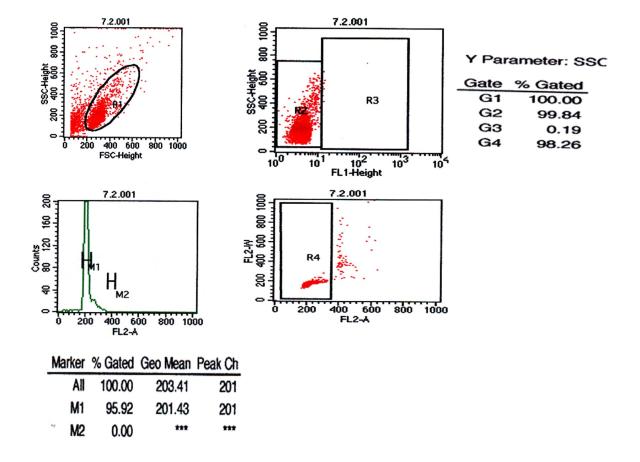 图片附件: 94994765.snap.jpg (2012-1-28 09:58, 39.89 KB) / 该附件被下载次数 8
图片附件: 94994765.snap.jpg (2012-1-28 09:58, 39.89 KB) / 该附件被下载次数 8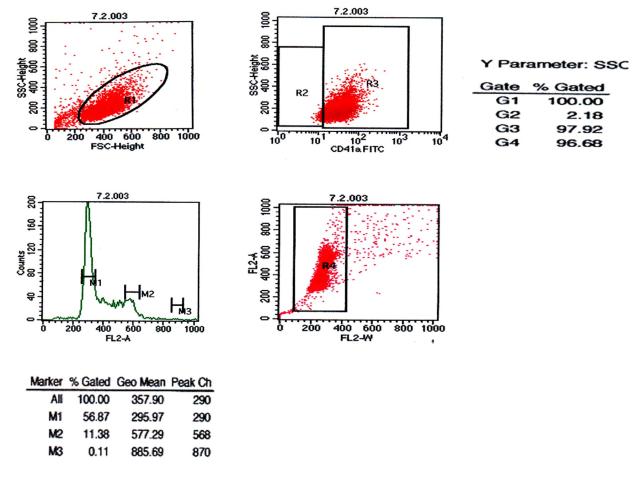 图片附件: 95896855.jpg (2012-1-28 10:00, 47.46 KB) / 该附件被下载次数 4
图片附件: 95896855.jpg (2012-1-28 10:00, 47.46 KB) / 该附件被下载次数 4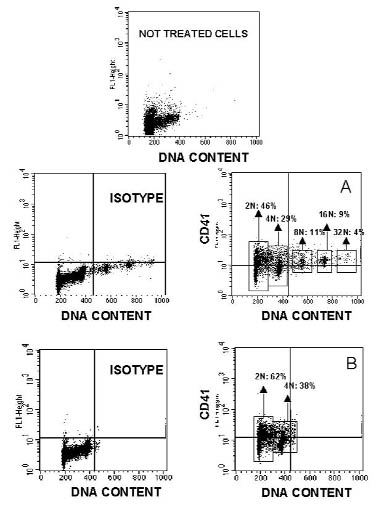 图片附件: 37210603.jpg (2012-1-28 10:01, 37.13 KB) / 该附件被下载次数 5
图片附件: 37210603.jpg (2012-1-28 10:01, 37.13 KB) / 该附件被下载次数 5 图片附件: 92602593.snap.jpg (2012-1-28 10:02, 19.51 KB) / 该附件被下载次数 6
图片附件: 92602593.snap.jpg (2012-1-28 10:02, 19.51 KB) / 该附件被下载次数 6 图片附件: 38838132.snap.jpg (2012-1-28 10:02, 17.57 KB) / 该附件被下载次数 6
图片附件: 38838132.snap.jpg (2012-1-28 10:02, 17.57 KB) / 该附件被下载次数 6 图片附件: 89316716.jpg (2012-1-28 10:06, 42.79 KB) / 该附件被下载次数 5
图片附件: 89316716.jpg (2012-1-28 10:06, 42.79 KB) / 该附件被下载次数 5 图片附件: 72016721.jpg (2012-1-28 10:07, 49.39 KB) / 该附件被下载次数 6
图片附件: 72016721.jpg (2012-1-28 10:07, 49.39 KB) / 该附件被下载次数 6 图片附件: 43356205.snap.jpg (2012-1-28 14:46, 56.42 KB) / 该附件被下载次数 5
图片附件: 43356205.snap.jpg (2012-1-28 14:46, 56.42 KB) / 该附件被下载次数 5 图片附件: 44794880.jpg (2012-1-28 15:32, 37.64 KB) / 该附件被下载次数 8
图片附件: 44794880.jpg (2012-1-28 15:32, 37.64 KB) / 该附件被下载次数 8 图片附件: 51908931.jpg (2012-1-28 15:33, 38.21 KB) / 该附件被下载次数 4
图片附件: 51908931.jpg (2012-1-28 15:33, 38.21 KB) / 该附件被下载次数 4 图片附件: 35022398.snap.jpg (2012-1-28 15:58, 32.78 KB) / 该附件被下载次数 4
图片附件: 35022398.snap.jpg (2012-1-28 15:58, 32.78 KB) / 该附件被下载次数 4 图片附件: 64581421.jpg (2012-1-28 16:07, 41.88 KB) / 该附件被下载次数 6
图片附件: 64581421.jpg (2012-1-28 16:07, 41.88 KB) / 该附件被下载次数 6 图片附件: 42952397.snap.jpg (2012-1-28 16:10, 96.04 KB) / 该附件被下载次数 5
图片附件: 42952397.snap.jpg (2012-1-28 16:10, 96.04 KB) / 该附件被下载次数 5 图片附件: 41055135.gif (2012-1-28 16:26, 8.68 KB) / 该附件被下载次数 4
图片附件: 41055135.gif (2012-1-28 16:26, 8.68 KB) / 该附件被下载次数 4 图片附件: 84325468.jpg (2012-1-28 17:27, 59.58 KB) / 该附件被下载次数 9
图片附件: 84325468.jpg (2012-1-28 17:27, 59.58 KB) / 该附件被下载次数 9 图片附件: 64575820.jpg (2012-1-28 17:38, 47.72 KB) / 该附件被下载次数 4
图片附件: 64575820.jpg (2012-1-28 17:38, 47.72 KB) / 该附件被下载次数 4 图片附件: 52542200.jpg (2012-1-28 17:38, 40.94 KB) / 该附件被下载次数 5
图片附件: 52542200.jpg (2012-1-28 17:38, 40.94 KB) / 该附件被下载次数 5 图片附件: 58739717.jpg (2012-1-29 08:35, 16.11 KB) / 该附件被下载次数 7
图片附件: 58739717.jpg (2012-1-29 08:35, 16.11 KB) / 该附件被下载次数 7 图片附件: 74177738.jpg (2012-1-29 08:36, 18.46 KB) / 该附件被下载次数 6
图片附件: 74177738.jpg (2012-1-29 08:36, 18.46 KB) / 该附件被下载次数 6 图片附件: 96562228.jpg (2012-1-29 08:36, 23.23 KB) / 该附件被下载次数 9
图片附件: 96562228.jpg (2012-1-29 08:36, 23.23 KB) / 该附件被下载次数 9 图片附件: 61077196.jpg (2012-1-29 08:36, 20.7 KB) / 该附件被下载次数 12
图片附件: 61077196.jpg (2012-1-29 08:36, 20.7 KB) / 该附件被下载次数 12 图片附件: 98996927.jpg (2012-1-29 08:37, 16.29 KB) / 该附件被下载次数 6
图片附件: 98996927.jpg (2012-1-29 08:37, 16.29 KB) / 该附件被下载次数 6 图片附件: 61498514.jpg (2012-1-29 08:37, 23.35 KB) / 该附件被下载次数 8
图片附件: 61498514.jpg (2012-1-29 08:37, 23.35 KB) / 该附件被下载次数 8
 图片附件: 20731232.snap.jpg (2012-1-29 09:28, 47.99 KB) / 该附件被下载次数 11
图片附件: 20731232.snap.jpg (2012-1-29 09:28, 47.99 KB) / 该附件被下载次数 11 图片附件: 30584731.snap.jpg (2012-1-29 09:29, 47.99 KB) / 该附件被下载次数 5
图片附件: 30584731.snap.jpg (2012-1-29 09:29, 47.99 KB) / 该附件被下载次数 5 图片附件: 58317360.snap.jpg (2012-1-29 10:23, 116.48 KB) / 该附件被下载次数 14
图片附件: 58317360.snap.jpg (2012-1-29 10:23, 116.48 KB) / 该附件被下载次数 14 图片附件: 53477449.snap.jpg (2012-1-29 10:40, 26.48 KB) / 该附件被下载次数 10
图片附件: 53477449.snap.jpg (2012-1-29 10:40, 26.48 KB) / 该附件被下载次数 10 图片附件: 67370558.jpg (2012-1-29 10:44, 40.21 KB) / 该附件被下载次数 10
图片附件: 67370558.jpg (2012-1-29 10:44, 40.21 KB) / 该附件被下载次数 10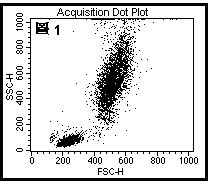 图片附件: 26874215.jpg (2012-1-29 10:44, 41.74 KB) / 该附件被下载次数 8
图片附件: 26874215.jpg (2012-1-29 10:44, 41.74 KB) / 该附件被下载次数 8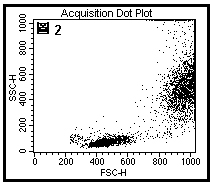 图片附件: 75188051.jpg (2012-1-29 10:45, 43.47 KB) / 该附件被下载次数 11
图片附件: 75188051.jpg (2012-1-29 10:45, 43.47 KB) / 该附件被下载次数 11 图片附件: 32608983.jpg (2012-1-29 10:45, 42.86 KB) / 该附件被下载次数 14
图片附件: 32608983.jpg (2012-1-29 10:45, 42.86 KB) / 该附件被下载次数 14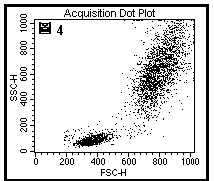 图片附件: 34969195.jpg (2012-1-29 11:37, 30.67 KB) / 该附件被下载次数 9
图片附件: 34969195.jpg (2012-1-29 11:37, 30.67 KB) / 该附件被下载次数 9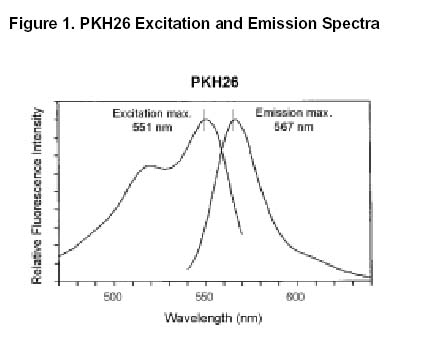 图片附件: 67752476.jpg (2012-1-29 11:42, 36.2 KB) / 该附件被下载次数 8
图片附件: 67752476.jpg (2012-1-29 11:42, 36.2 KB) / 该附件被下载次数 8 图片附件: 56511466.jpg (2012-1-29 12:02, 52.22 KB) / 该附件被下载次数 9
图片附件: 56511466.jpg (2012-1-29 12:02, 52.22 KB) / 该附件被下载次数 9 图片附件: 86584741.jpg (2012-1-29 12:02, 52.27 KB) / 该附件被下载次数 6
图片附件: 86584741.jpg (2012-1-29 12:02, 52.27 KB) / 该附件被下载次数 6 图片附件: 34566939.jpg (2012-1-29 12:03, 48.55 KB) / 该附件被下载次数 8
图片附件: 34566939.jpg (2012-1-29 12:03, 48.55 KB) / 该附件被下载次数 8 图片附件: 21257839.jpg (2012-1-29 13:12, 25.54 KB) / 该附件被下载次数 7
图片附件: 21257839.jpg (2012-1-29 13:12, 25.54 KB) / 该附件被下载次数 7 图片附件: 52400969.jpg (2012-1-29 15:11, 13.38 KB) / 该附件被下载次数 6
图片附件: 52400969.jpg (2012-1-29 15:11, 13.38 KB) / 该附件被下载次数 6 图片附件: 39432428.jpg (2012-1-29 15:12, 10.27 KB) / 该附件被下载次数 4
图片附件: 39432428.jpg (2012-1-29 15:12, 10.27 KB) / 该附件被下载次数 4 图片附件: 66324586.jpg (2012-1-29 15:12, 12.07 KB) / 该附件被下载次数 6
图片附件: 66324586.jpg (2012-1-29 15:12, 12.07 KB) / 该附件被下载次数 6 图片附件: 28553072.jpg (2012-1-29 15:14, 23.09 KB) / 该附件被下载次数 7
图片附件: 28553072.jpg (2012-1-29 15:14, 23.09 KB) / 该附件被下载次数 7 图片附件: 50747564.jpg (2012-1-29 15:14, 37.81 KB) / 该附件被下载次数 9
图片附件: 50747564.jpg (2012-1-29 15:14, 37.81 KB) / 该附件被下载次数 9 图片附件: 68385067.snap.jpg (2012-1-29 15:15, 27.59 KB) / 该附件被下载次数 6
图片附件: 68385067.snap.jpg (2012-1-29 15:15, 27.59 KB) / 该附件被下载次数 6 图片附件: 25050570.snap.jpg (2012-1-29 15:16, 50.55 KB) / 该附件被下载次数 7
图片附件: 25050570.snap.jpg (2012-1-29 15:16, 50.55 KB) / 该附件被下载次数 7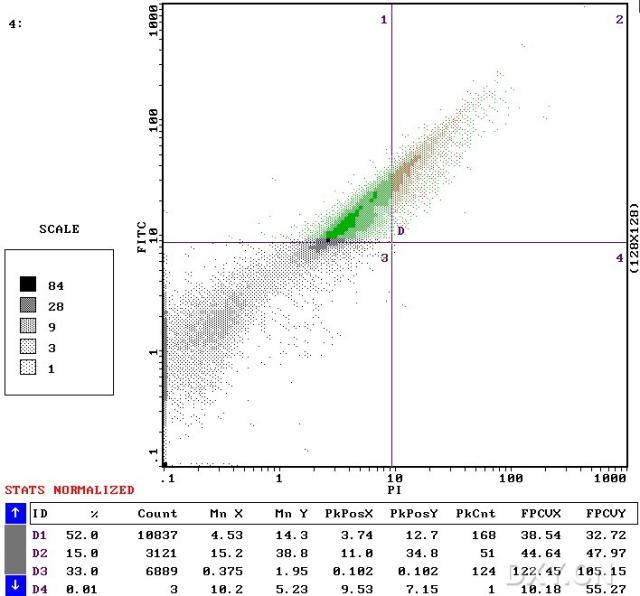 图片附件: 12063691.jpg (2012-1-29 15:35, 35.01 KB) / 该附件被下载次数 7
图片附件: 12063691.jpg (2012-1-29 15:35, 35.01 KB) / 该附件被下载次数 7 图片附件: 61965839.jpg (2012-1-29 15:35, 25.04 KB) / 该附件被下载次数 9
图片附件: 61965839.jpg (2012-1-29 15:35, 25.04 KB) / 该附件被下载次数 9 图片附件: 37772655.jpg (2012-1-29 15:51, 8.15 KB) / 该附件被下载次数 9
图片附件: 37772655.jpg (2012-1-29 15:51, 8.15 KB) / 该附件被下载次数 9 图片附件: 67771287.jpg (2012-1-29 16:18, 58.82 KB) / 该附件被下载次数 5
图片附件: 67771287.jpg (2012-1-29 16:18, 58.82 KB) / 该附件被下载次数 5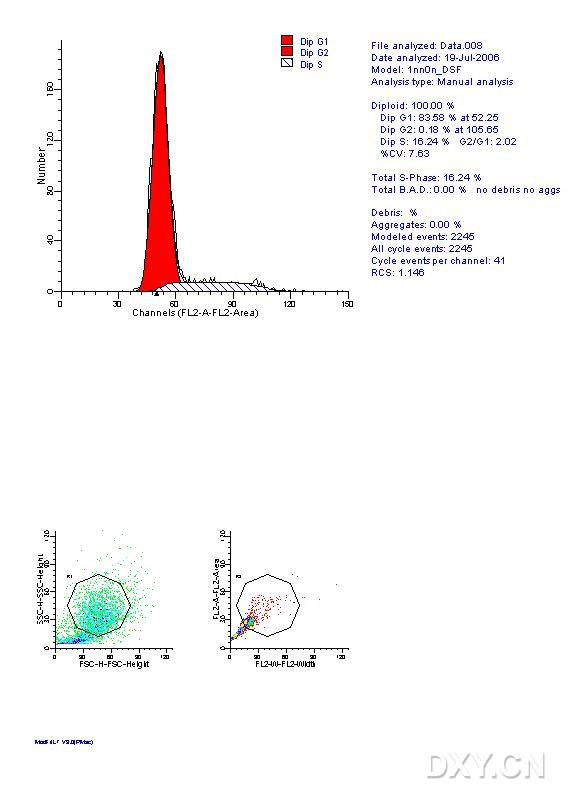 图片附件: 38359778.jpg (2012-1-29 16:37, 12.81 KB) / 该附件被下载次数 8
图片附件: 38359778.jpg (2012-1-29 16:37, 12.81 KB) / 该附件被下载次数 8 图片附件: 36803514.snap.jpg (2012-1-29 16:46, 37.64 KB) / 该附件被下载次数 7
图片附件: 36803514.snap.jpg (2012-1-29 16:46, 37.64 KB) / 该附件被下载次数 7 图片附件: 18981560.png (2012-1-29 16:52, 24.44 KB) / 该附件被下载次数 7
图片附件: 18981560.png (2012-1-29 16:52, 24.44 KB) / 该附件被下载次数 7 图片附件: 33906718.png (2012-1-29 16:52, 57.09 KB) / 该附件被下载次数 7
图片附件: 33906718.png (2012-1-29 16:52, 57.09 KB) / 该附件被下载次数 7 图片附件: 91066791.png (2012-1-29 16:53, 55.27 KB) / 该附件被下载次数 8
图片附件: 91066791.png (2012-1-29 16:53, 55.27 KB) / 该附件被下载次数 8 图片附件: 79674750.jpg (2012-1-30 14:22, 20.33 KB) / 该附件被下载次数 8
图片附件: 79674750.jpg (2012-1-30 14:22, 20.33 KB) / 该附件被下载次数 8 图片附件: 98021569.jpg (2012-1-30 14:36, 32.8 KB) / 该附件被下载次数 8
图片附件: 98021569.jpg (2012-1-30 14:36, 32.8 KB) / 该附件被下载次数 8 图片附件: 16448321.jpg (2012-1-30 14:36, 33.08 KB) / 该附件被下载次数 8
图片附件: 16448321.jpg (2012-1-30 14:36, 33.08 KB) / 该附件被下载次数 8 图片附件: 94646919.jpg (2012-1-30 14:46, 33.08 KB) / 该附件被下载次数 9
图片附件: 94646919.jpg (2012-1-30 14:46, 33.08 KB) / 该附件被下载次数 9 图片附件: 67002305.snap.jpg (2012-1-30 14:57, 67.76 KB) / 该附件被下载次数 9
图片附件: 67002305.snap.jpg (2012-1-30 14:57, 67.76 KB) / 该附件被下载次数 9 图片附件: 85677441.snap.jpg (2012-1-30 14:57, 90.8 KB) / 该附件被下载次数 10
图片附件: 85677441.snap.jpg (2012-1-30 14:57, 90.8 KB) / 该附件被下载次数 10 图片附件: 18846492.snap.jpg (2012-1-30 14:58, 72.58 KB) / 该附件被下载次数 6
图片附件: 18846492.snap.jpg (2012-1-30 14:58, 72.58 KB) / 该附件被下载次数 6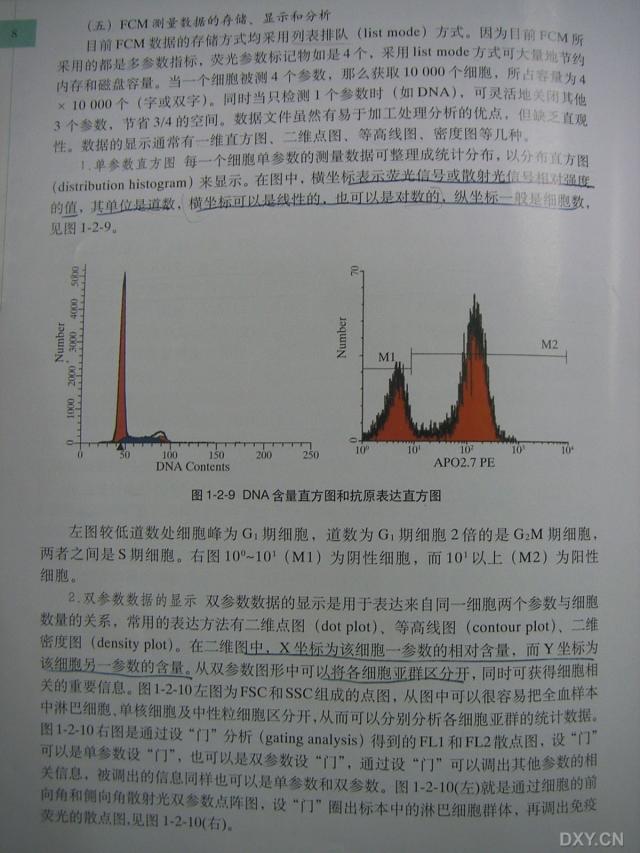 图片附件: 61032680.snap.jpg (2012-1-30 14:58, 48.13 KB) / 该附件被下载次数 8
图片附件: 61032680.snap.jpg (2012-1-30 14:58, 48.13 KB) / 该附件被下载次数 8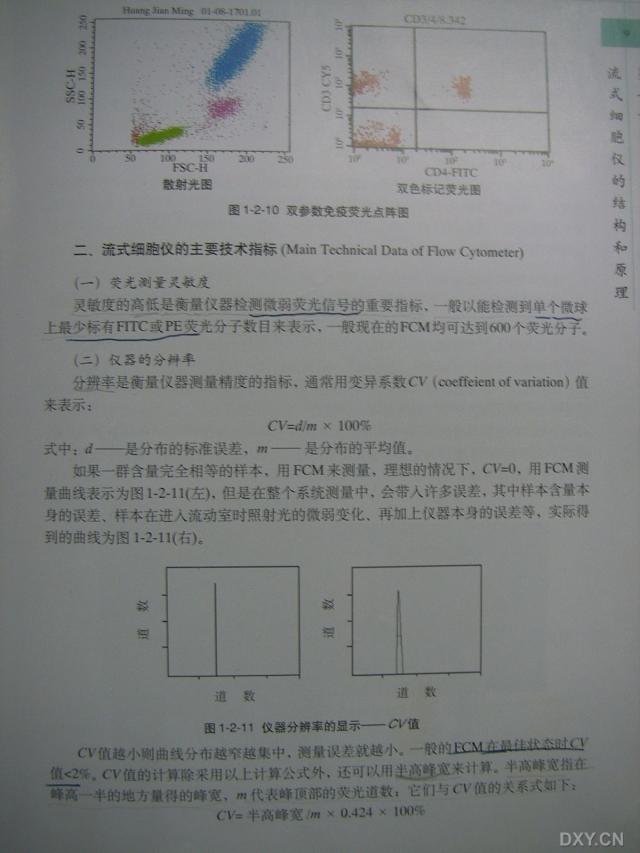 图片附件: 49949339.snap.jpg (2012-1-30 14:59, 61.07 KB) / 该附件被下载次数 5
图片附件: 49949339.snap.jpg (2012-1-30 14:59, 61.07 KB) / 该附件被下载次数 5 图片附件: 71326785.snap.jpg (2012-1-30 14:59, 49.98 KB) / 该附件被下载次数 6
图片附件: 71326785.snap.jpg (2012-1-30 14:59, 49.98 KB) / 该附件被下载次数 6 图片附件: 97349892.snap.jpg (2012-1-30 15:00, 59.57 KB) / 该附件被下载次数 7
图片附件: 97349892.snap.jpg (2012-1-30 15:00, 59.57 KB) / 该附件被下载次数 7 图片附件: 45584651.snap.jpg (2012-1-30 15:00, 56.81 KB) / 该附件被下载次数 7
图片附件: 45584651.snap.jpg (2012-1-30 15:00, 56.81 KB) / 该附件被下载次数 7 图片附件: 90444276.snap.jpg (2012-1-30 15:00, 77.44 KB) / 该附件被下载次数 7
图片附件: 90444276.snap.jpg (2012-1-30 15:00, 77.44 KB) / 该附件被下载次数 7 图片附件: 91302060.snap.jpg (2012-1-30 15:01, 72.49 KB) / 该附件被下载次数 8
图片附件: 91302060.snap.jpg (2012-1-30 15:01, 72.49 KB) / 该附件被下载次数 8 图片附件: 78196464.snap.jpg (2012-1-30 15:01, 69.11 KB) / 该附件被下载次数 8
图片附件: 78196464.snap.jpg (2012-1-30 15:01, 69.11 KB) / 该附件被下载次数 8 图片附件: 71132043.snap.jpg (2012-1-30 15:01, 62.29 KB) / 该附件被下载次数 6
图片附件: 71132043.snap.jpg (2012-1-30 15:01, 62.29 KB) / 该附件被下载次数 6 图片附件: 91940452.snap.jpg (2012-1-30 15:02, 54.86 KB) / 该附件被下载次数 9
图片附件: 91940452.snap.jpg (2012-1-30 15:02, 54.86 KB) / 该附件被下载次数 9 图片附件: 47291962.snap.jpg (2012-1-30 15:02, 94.13 KB) / 该附件被下载次数 6
图片附件: 47291962.snap.jpg (2012-1-30 15:02, 94.13 KB) / 该附件被下载次数 6 图片附件: 91383848.snap.jpg (2012-1-30 15:04, 72.66 KB) / 该附件被下载次数 6
图片附件: 91383848.snap.jpg (2012-1-30 15:04, 72.66 KB) / 该附件被下载次数 6 图片附件: 54409215.snap.jpg (2012-1-30 15:04, 70.22 KB) / 该附件被下载次数 6
图片附件: 54409215.snap.jpg (2012-1-30 15:04, 70.22 KB) / 该附件被下载次数 6 图片附件: 56797820.snap.jpg (2012-1-30 15:05, 68.24 KB) / 该附件被下载次数 7
图片附件: 56797820.snap.jpg (2012-1-30 15:05, 68.24 KB) / 该附件被下载次数 7 图片附件: 10295278.snap.jpg (2012-1-30 15:05, 73.86 KB) / 该附件被下载次数 8
图片附件: 10295278.snap.jpg (2012-1-30 15:05, 73.86 KB) / 该附件被下载次数 8 图片附件: 10618914.snap.jpg (2012-1-30 15:06, 72.52 KB) / 该附件被下载次数 7
图片附件: 10618914.snap.jpg (2012-1-30 15:06, 72.52 KB) / 该附件被下载次数 7 图片附件: 61152364.snap.jpg (2012-1-30 15:06, 69.55 KB) / 该附件被下载次数 9
图片附件: 61152364.snap.jpg (2012-1-30 15:06, 69.55 KB) / 该附件被下载次数 9 图片附件: 26992361.snap.jpg (2012-1-30 15:07, 70.8 KB) / 该附件被下载次数 6
图片附件: 26992361.snap.jpg (2012-1-30 15:07, 70.8 KB) / 该附件被下载次数 6 图片附件: 26992361.snap.jpg (2012-1-30 15:11, 70.8 KB) / 该附件被下载次数 124
图片附件: 26992361.snap.jpg (2012-1-30 15:11, 70.8 KB) / 该附件被下载次数 124 图片附件: 74511348.jpg (2012-1-30 15:11, 20.49 KB) / 该附件被下载次数 6
图片附件: 74511348.jpg (2012-1-30 15:11, 20.49 KB) / 该附件被下载次数 6 图片附件: 23405108.jpg (2012-1-30 15:11, 20.54 KB) / 该附件被下载次数 13
图片附件: 23405108.jpg (2012-1-30 15:11, 20.54 KB) / 该附件被下载次数 13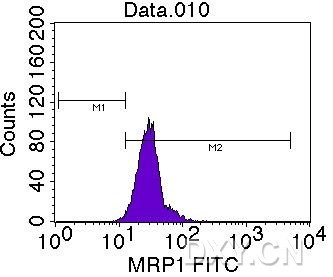 图片附件: 10782481.snap.jpg (2012-1-30 15:49, 30.81 KB) / 该附件被下载次数 6
图片附件: 10782481.snap.jpg (2012-1-30 15:49, 30.81 KB) / 该附件被下载次数 6 图片附件: 30164061.snap.jpg (2012-1-30 15:50, 12.65 KB) / 该附件被下载次数 5
图片附件: 30164061.snap.jpg (2012-1-30 15:50, 12.65 KB) / 该附件被下载次数 5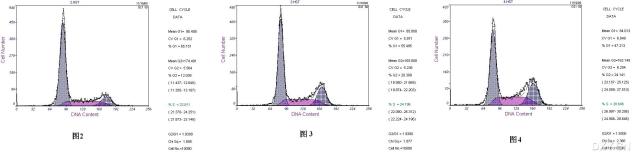 图片附件: 48354867.snap.jpg (2012-1-30 16:11, 27.36 KB) / 该附件被下载次数 12
图片附件: 48354867.snap.jpg (2012-1-30 16:11, 27.36 KB) / 该附件被下载次数 12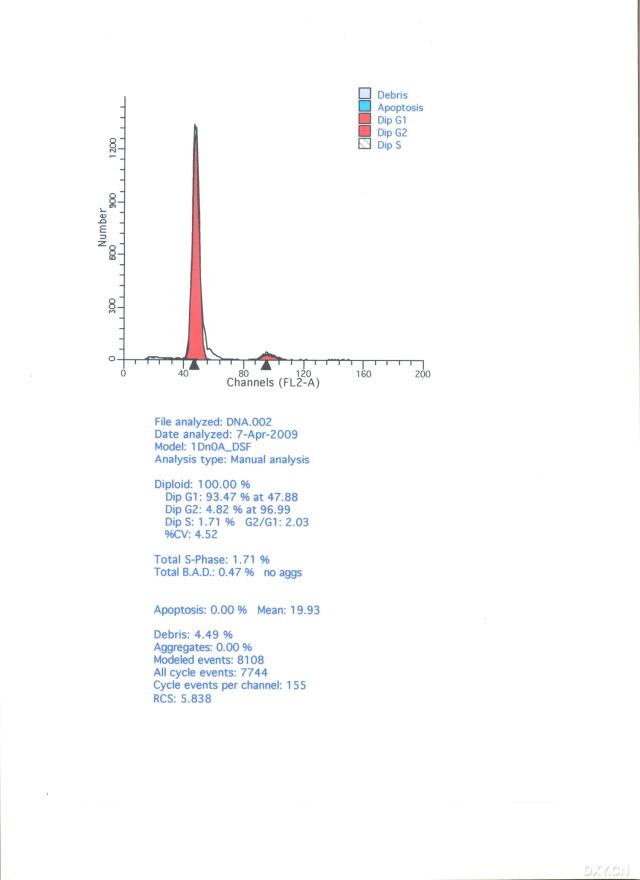 图片附件: 51731431.snap.jpg (2012-1-30 16:21, 21.7 KB) / 该附件被下载次数 6
图片附件: 51731431.snap.jpg (2012-1-30 16:21, 21.7 KB) / 该附件被下载次数 6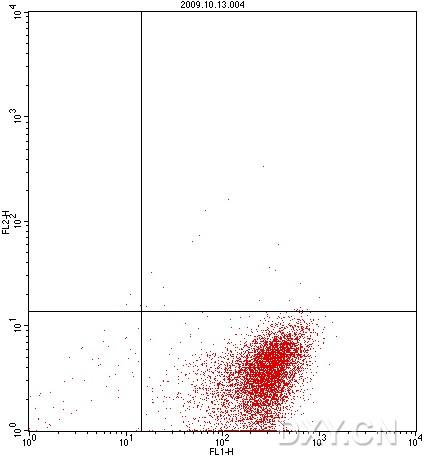 图片附件: 31717192.snap.jpg (2012-1-30 16:23, 51.64 KB) / 该附件被下载次数 5
图片附件: 31717192.snap.jpg (2012-1-30 16:23, 51.64 KB) / 该附件被下载次数 5 图片附件: 48611389.snap.jpg (2012-1-30 16:35, 52.11 KB) / 该附件被下载次数 4
图片附件: 48611389.snap.jpg (2012-1-30 16:35, 52.11 KB) / 该附件被下载次数 4 图片附件: 23250859.snap.jpg (2012-1-30 16:36, 53.3 KB) / 该附件被下载次数 10
图片附件: 23250859.snap.jpg (2012-1-30 16:36, 53.3 KB) / 该附件被下载次数 10 图片附件: 64351359.snap.jpg (2012-1-30 16:41, 40.39 KB) / 该附件被下载次数 4
图片附件: 64351359.snap.jpg (2012-1-30 16:41, 40.39 KB) / 该附件被下载次数 4 图片附件: 29902504.png (2012-1-30 16:43, 8.36 KB) / 该附件被下载次数 8
图片附件: 29902504.png (2012-1-30 16:43, 8.36 KB) / 该附件被下载次数 8 图片附件: 55138923.jpg (2012-1-30 16:44, 16.77 KB) / 该附件被下载次数 5
图片附件: 55138923.jpg (2012-1-30 16:44, 16.77 KB) / 该附件被下载次数 5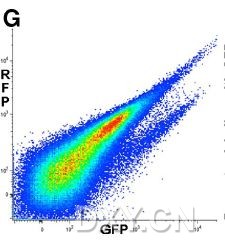 图片附件: 38138099.jpg (2012-1-30 16:45, 14.27 KB) / 该附件被下载次数 6
图片附件: 38138099.jpg (2012-1-30 16:45, 14.27 KB) / 该附件被下载次数 6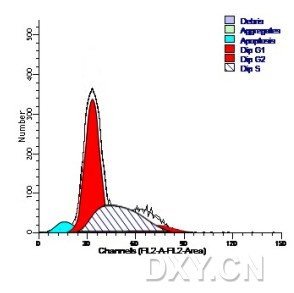 图片附件: 13253261.jpg (2012-1-30 17:45, 43.29 KB) / 该附件被下载次数 10
图片附件: 13253261.jpg (2012-1-30 17:45, 43.29 KB) / 该附件被下载次数 10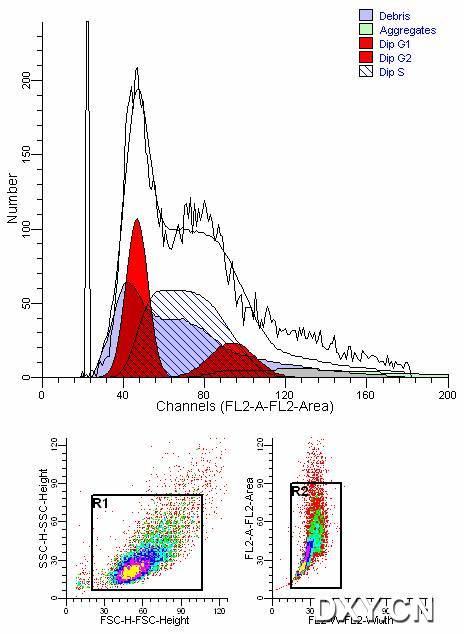 图片附件: 15771239.jpg (2012-1-31 08:45, 52.22 KB) / 该附件被下载次数 11
图片附件: 15771239.jpg (2012-1-31 08:45, 52.22 KB) / 该附件被下载次数 11 图片附件: 67335153.jpg (2012-1-31 08:45, 52.27 KB) / 该附件被下载次数 6
图片附件: 67335153.jpg (2012-1-31 08:45, 52.27 KB) / 该附件被下载次数 6 图片附件: 63551530.jpg (2012-1-31 08:45, 48.55 KB) / 该附件被下载次数 7
图片附件: 63551530.jpg (2012-1-31 08:45, 48.55 KB) / 该附件被下载次数 7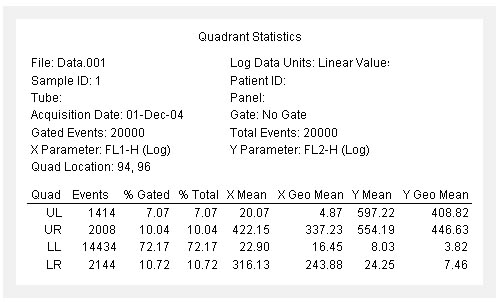 图片附件: 90653975.jpg (2012-1-31 08:47, 59.98 KB) / 该附件被下载次数 6
图片附件: 90653975.jpg (2012-1-31 08:47, 59.98 KB) / 该附件被下载次数 6 图片附件: 68176125.jpg (2012-1-31 08:48, 55.77 KB) / 该附件被下载次数 6
图片附件: 68176125.jpg (2012-1-31 08:48, 55.77 KB) / 该附件被下载次数 6 图片附件: 32264918.jpg (2012-1-31 10:26, 37.61 KB) / 该附件被下载次数 5
图片附件: 32264918.jpg (2012-1-31 10:26, 37.61 KB) / 该附件被下载次数 5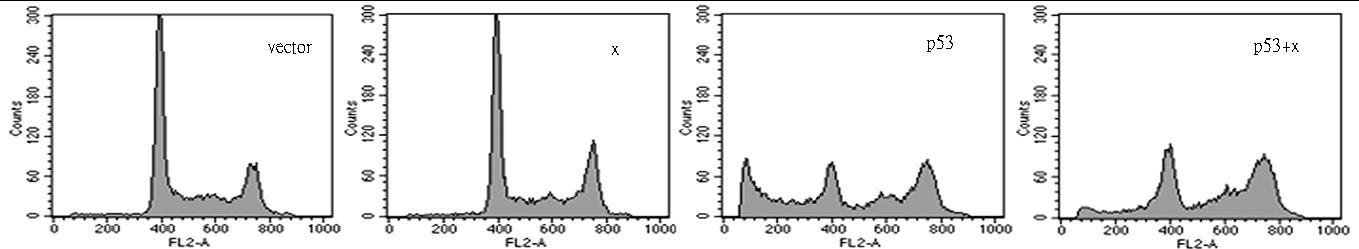 图片附件: 51277831.jpg (2012-1-31 10:27, 46.15 KB) / 该附件被下载次数 11
图片附件: 51277831.jpg (2012-1-31 10:27, 46.15 KB) / 该附件被下载次数 11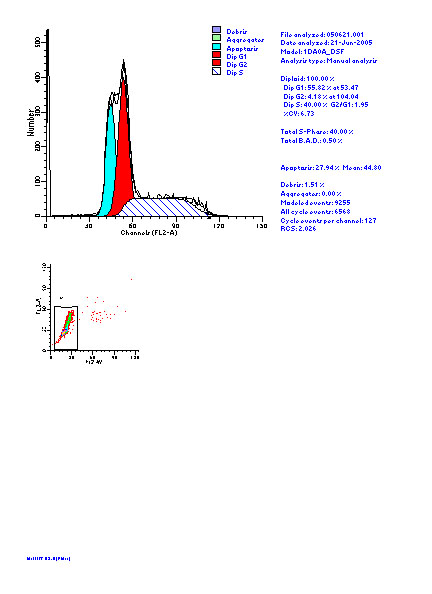 图片附件: 54824790.jpg (2012-1-31 10:27, 56.07 KB) / 该附件被下载次数 7
图片附件: 54824790.jpg (2012-1-31 10:27, 56.07 KB) / 该附件被下载次数 7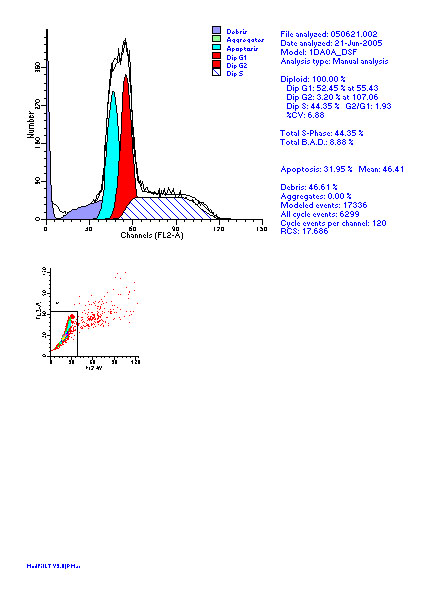 图片附件: 78501733.jpg (2012-1-31 10:28, 54.79 KB) / 该附件被下载次数 7
图片附件: 78501733.jpg (2012-1-31 10:28, 54.79 KB) / 该附件被下载次数 7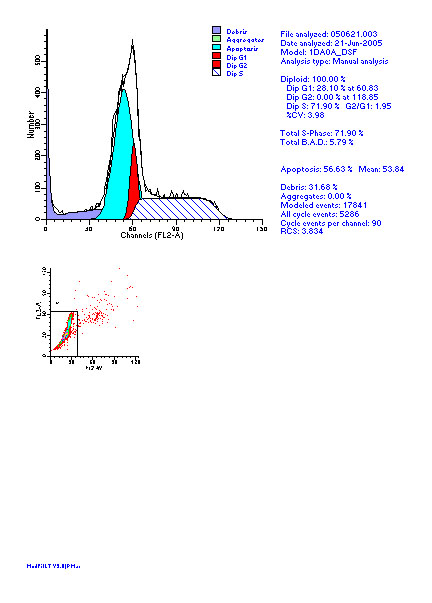 图片附件: 91901908.jpg (2012-1-31 10:33, 24.15 KB) / 该附件被下载次数 6
图片附件: 91901908.jpg (2012-1-31 10:33, 24.15 KB) / 该附件被下载次数 6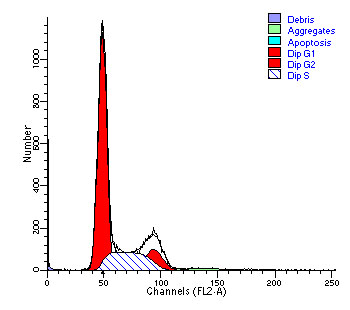 图片附件: 28435554.jpg (2012-1-31 10:34, 24.51 KB) / 该附件被下载次数 3
图片附件: 28435554.jpg (2012-1-31 10:34, 24.51 KB) / 该附件被下载次数 3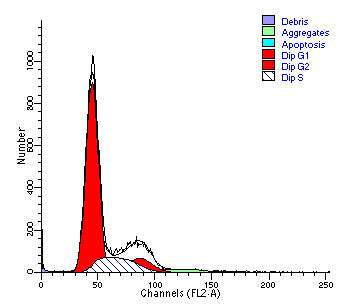 图片附件: 25795333.jpg (2012-1-31 10:34, 25.27 KB) / 该附件被下载次数 9
图片附件: 25795333.jpg (2012-1-31 10:34, 25.27 KB) / 该附件被下载次数 9 图片附件: 82595115.jpg (2012-1-31 10:35, 21.52 KB) / 该附件被下载次数 6
图片附件: 82595115.jpg (2012-1-31 10:35, 21.52 KB) / 该附件被下载次数 6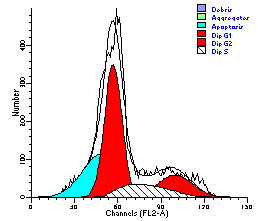 图片附件: 89085642.jpg (2012-1-31 10:35, 22.53 KB) / 该附件被下载次数 6
图片附件: 89085642.jpg (2012-1-31 10:35, 22.53 KB) / 该附件被下载次数 6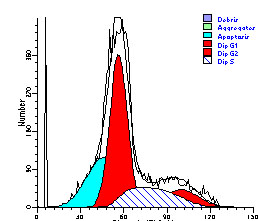 图片附件: 13612287.jpg (2012-1-31 10:37, 20.81 KB) / 该附件被下载次数 12
图片附件: 13612287.jpg (2012-1-31 10:37, 20.81 KB) / 该附件被下载次数 12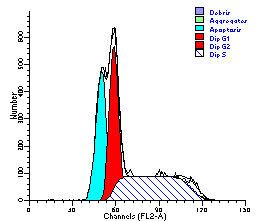 图片附件: 16528679.jpg (2012-1-31 10:39, 20.65 KB) / 该附件被下载次数 7
图片附件: 16528679.jpg (2012-1-31 10:39, 20.65 KB) / 该附件被下载次数 7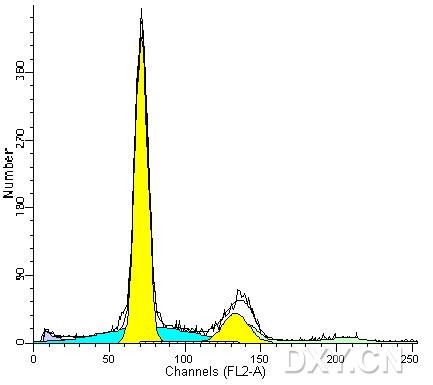 图片附件: 85564101.jpg (2012-1-31 10:40, 20.07 KB) / 该附件被下载次数 8
图片附件: 85564101.jpg (2012-1-31 10:40, 20.07 KB) / 该附件被下载次数 8 图片附件: 99841584.jpg (2012-1-31 10:42, 11.95 KB) / 该附件被下载次数 10
图片附件: 99841584.jpg (2012-1-31 10:42, 11.95 KB) / 该附件被下载次数 10 图片附件: 98567258.jpg (2012-1-31 10:49, 16.9 KB) / 该附件被下载次数 6
图片附件: 98567258.jpg (2012-1-31 10:49, 16.9 KB) / 该附件被下载次数 6 图片附件: 20063542.jpg (2012-1-31 10:50, 40.61 KB) / 该附件被下载次数 7
图片附件: 20063542.jpg (2012-1-31 10:50, 40.61 KB) / 该附件被下载次数 7 图片附件: 90058775.jpg (2012-1-31 10:50, 15.67 KB) / 该附件被下载次数 9
图片附件: 90058775.jpg (2012-1-31 10:50, 15.67 KB) / 该附件被下载次数 9 图片附件: 41889803.jpg (2012-1-31 10:50, 15.95 KB) / 该附件被下载次数 7
图片附件: 41889803.jpg (2012-1-31 10:50, 15.95 KB) / 该附件被下载次数 7 图片附件: 70123958.gif (2012-1-31 10:54, 36.79 KB) / 该附件被下载次数 7
图片附件: 70123958.gif (2012-1-31 10:54, 36.79 KB) / 该附件被下载次数 7 图片附件: 99598917.jpg (2012-1-31 10:55, 47.04 KB) / 该附件被下载次数 5
图片附件: 99598917.jpg (2012-1-31 10:55, 47.04 KB) / 该附件被下载次数 5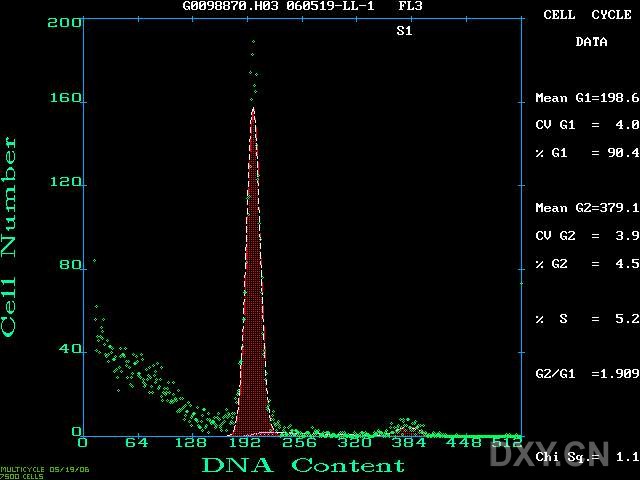 图片附件: 63471933.jpg (2012-1-31 10:57, 59.75 KB) / 该附件被下载次数 8
图片附件: 63471933.jpg (2012-1-31 10:57, 59.75 KB) / 该附件被下载次数 8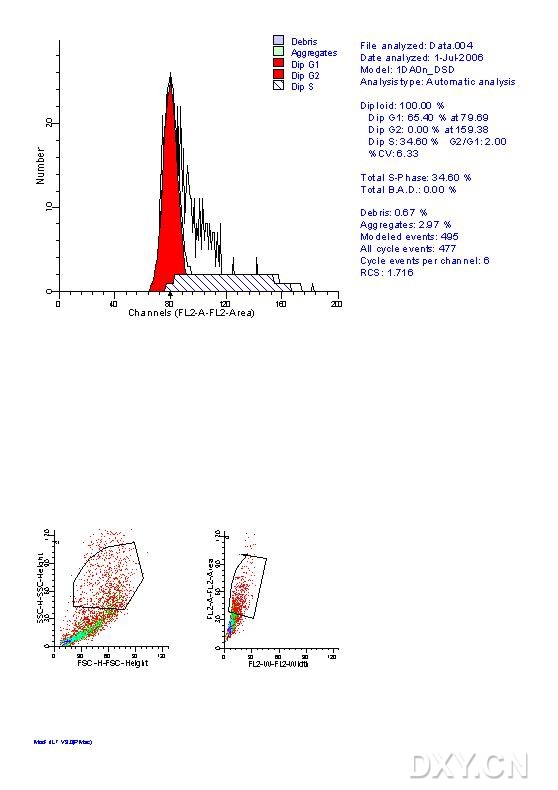 图片附件: 22006461.jpg (2012-1-31 10:57, 60.23 KB) / 该附件被下载次数 7
图片附件: 22006461.jpg (2012-1-31 10:57, 60.23 KB) / 该附件被下载次数 7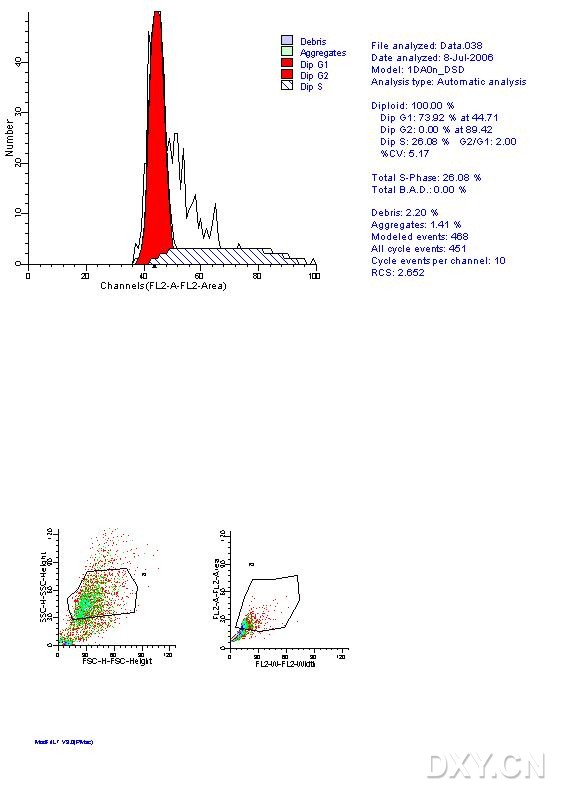 图片附件: 74178387.jpg (2012-1-31 10:58, 57.33 KB) / 该附件被下载次数 7
图片附件: 74178387.jpg (2012-1-31 10:58, 57.33 KB) / 该附件被下载次数 7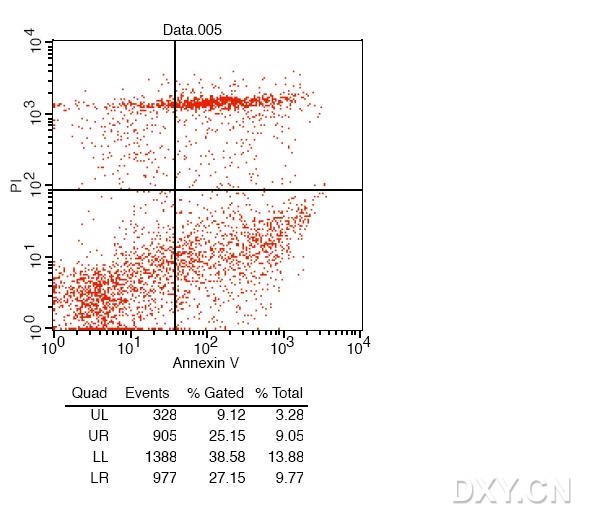 图片附件: 32986240.png (2012-1-31 11:41, 16.47 KB) / 该附件被下载次数 6
图片附件: 32986240.png (2012-1-31 11:41, 16.47 KB) / 该附件被下载次数 6 图片附件: 71534220.png (2012-1-31 11:41, 20.37 KB) / 该附件被下载次数 7
图片附件: 71534220.png (2012-1-31 11:41, 20.37 KB) / 该附件被下载次数 7 图片附件: 44781659.snap.jpg (2012-1-31 12:22, 43.87 KB) / 该附件被下载次数 6
图片附件: 44781659.snap.jpg (2012-1-31 12:22, 43.87 KB) / 该附件被下载次数 6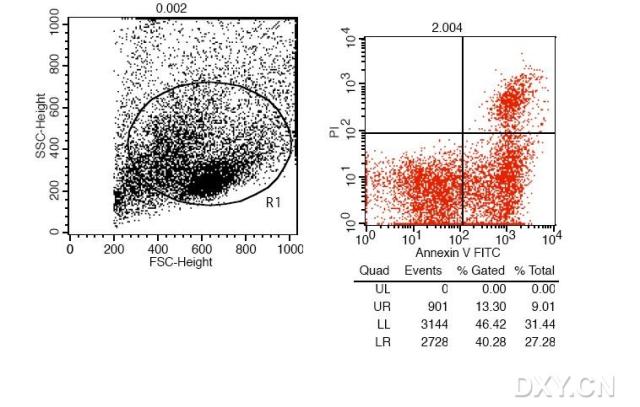 图片附件: 61063587.gif (2012-1-31 12:23, 13.12 KB) / 该附件被下载次数 7
图片附件: 61063587.gif (2012-1-31 12:23, 13.12 KB) / 该附件被下载次数 7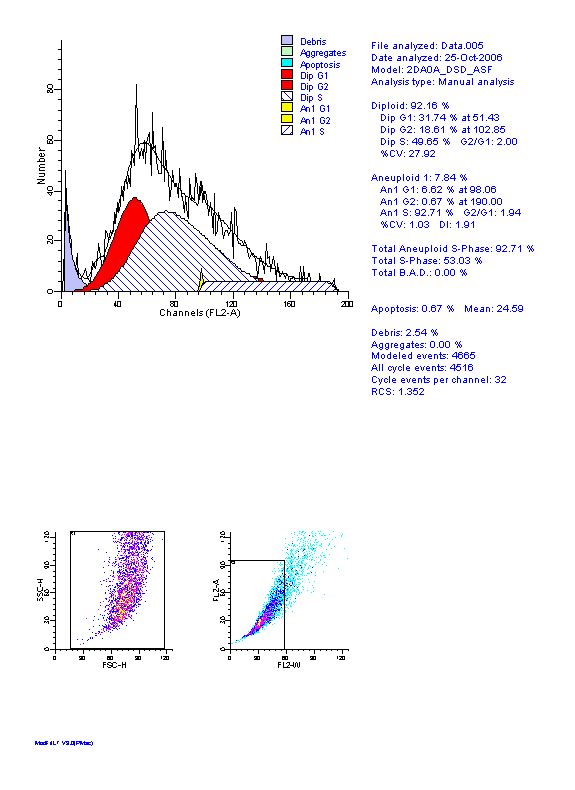 图片附件: 84406901.jpg (2012-1-31 12:26, 25.69 KB) / 该附件被下载次数 5
图片附件: 84406901.jpg (2012-1-31 12:26, 25.69 KB) / 该附件被下载次数 5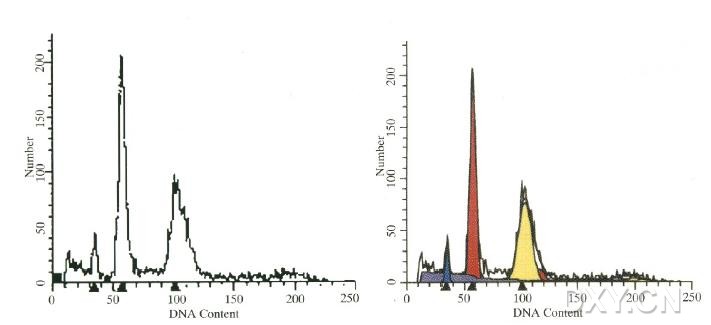 图片附件: 69536647.jpg (2012-1-31 12:27, 26.89 KB) / 该附件被下载次数 9
图片附件: 69536647.jpg (2012-1-31 12:27, 26.89 KB) / 该附件被下载次数 9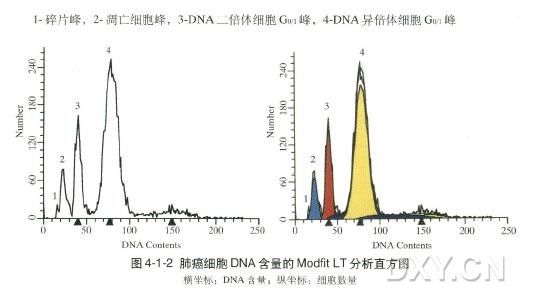 图片附件: 71963973.jpg (2012-1-31 12:27, 18.6 KB) / 该附件被下载次数 3
图片附件: 71963973.jpg (2012-1-31 12:27, 18.6 KB) / 该附件被下载次数 3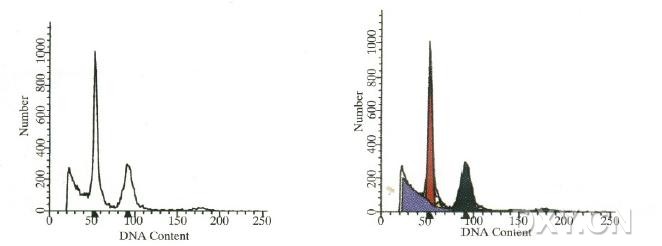 图片附件: 66293558.jpg (2012-1-31 12:28, 23.19 KB) / 该附件被下载次数 7
图片附件: 66293558.jpg (2012-1-31 12:28, 23.19 KB) / 该附件被下载次数 7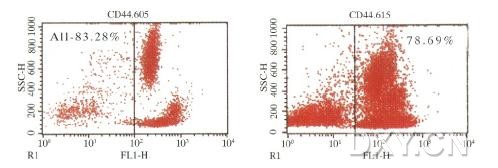 图片附件: 76436793.jpg (2012-1-31 12:28, 25.69 KB) / 该附件被下载次数 8
图片附件: 76436793.jpg (2012-1-31 12:28, 25.69 KB) / 该附件被下载次数 8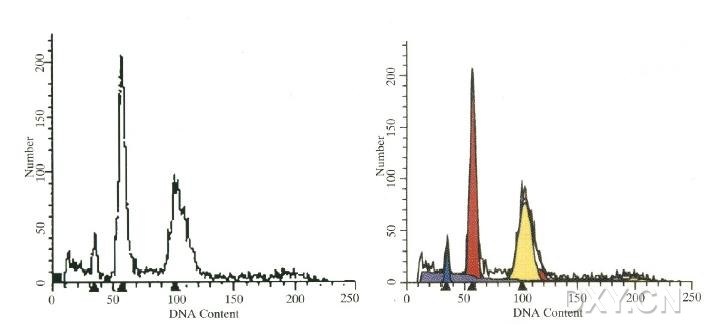 图片附件: 70974490.png (2012-1-31 12:31, 24.44 KB) / 该附件被下载次数 5
图片附件: 70974490.png (2012-1-31 12:31, 24.44 KB) / 该附件被下载次数 5 图片附件: 52364325.png (2012-1-31 12:32, 57.09 KB) / 该附件被下载次数 6
图片附件: 52364325.png (2012-1-31 12:32, 57.09 KB) / 该附件被下载次数 6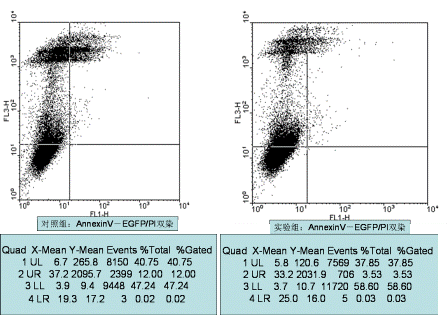 图片附件: 18409563.png (2012-1-31 12:32, 55.27 KB) / 该附件被下载次数 8
图片附件: 18409563.png (2012-1-31 12:32, 55.27 KB) / 该附件被下载次数 8 图片附件: 36189977.jpg (2012-1-31 12:33, 12.06 KB) / 该附件被下载次数 6
图片附件: 36189977.jpg (2012-1-31 12:33, 12.06 KB) / 该附件被下载次数 6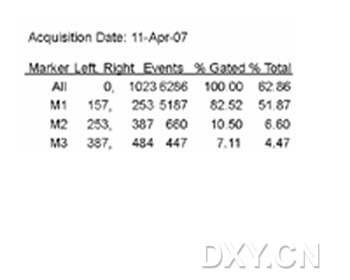 图片附件: 22728087.snap.jpg (2012-1-31 12:34, 30.96 KB) / 该附件被下载次数 6
图片附件: 22728087.snap.jpg (2012-1-31 12:34, 30.96 KB) / 该附件被下载次数 6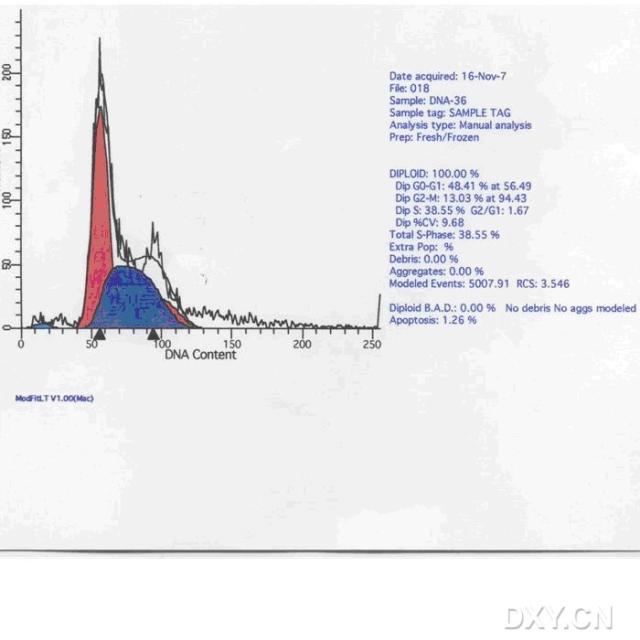 图片附件: 18612214.jpg (2012-1-31 12:35, 62.55 KB) / 该附件被下载次数 8
图片附件: 18612214.jpg (2012-1-31 12:35, 62.55 KB) / 该附件被下载次数 8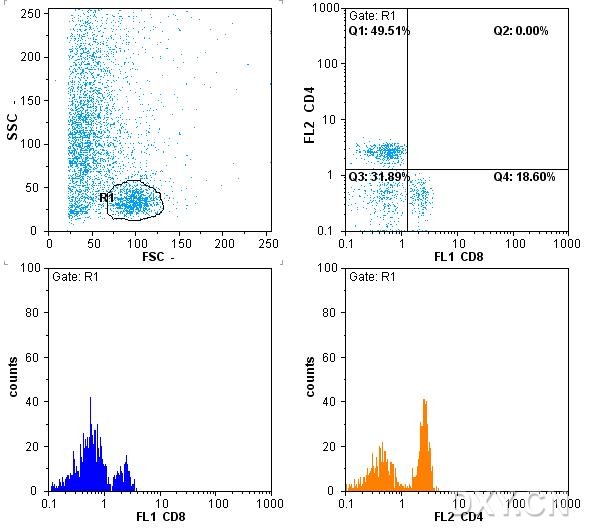 图片附件: 95875075.jpg (2012-1-31 12:36, 37.73 KB) / 该附件被下载次数 8
图片附件: 95875075.jpg (2012-1-31 12:36, 37.73 KB) / 该附件被下载次数 8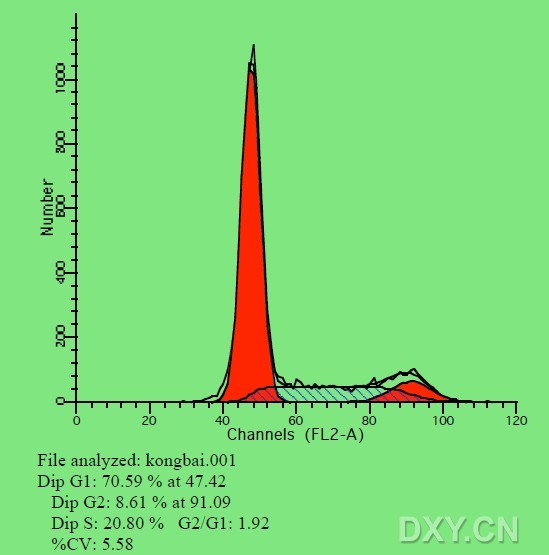 图片附件: 79095953.snap.jpg (2012-1-31 12:37, 42.1 KB) / 该附件被下载次数 5
图片附件: 79095953.snap.jpg (2012-1-31 12:37, 42.1 KB) / 该附件被下载次数 5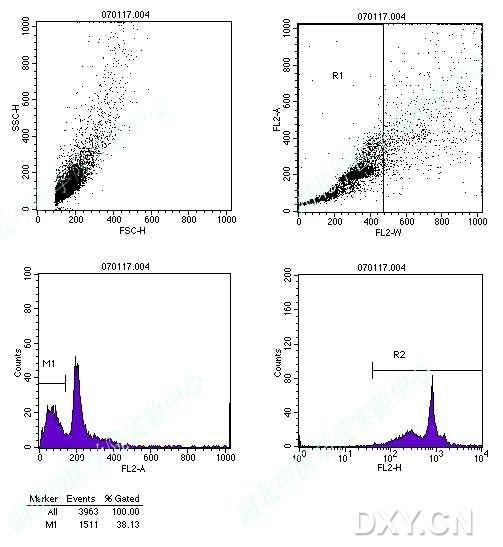 图片附件: 64422952.jpg (2012-1-31 12:39, 50.79 KB) / 该附件被下载次数 8
图片附件: 64422952.jpg (2012-1-31 12:39, 50.79 KB) / 该附件被下载次数 8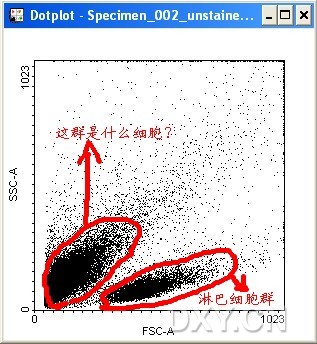 图片附件: 75483451.png (2012-1-31 12:39, 35.98 KB) / 该附件被下载次数 15
图片附件: 75483451.png (2012-1-31 12:39, 35.98 KB) / 该附件被下载次数 15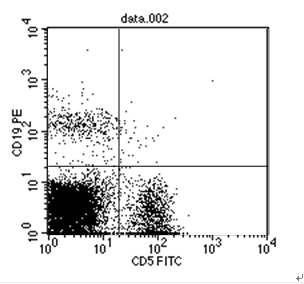 图片附件: 95276545.snap.jpg (2012-1-31 12:40, 35.87 KB) / 该附件被下载次数 8
图片附件: 95276545.snap.jpg (2012-1-31 12:40, 35.87 KB) / 该附件被下载次数 8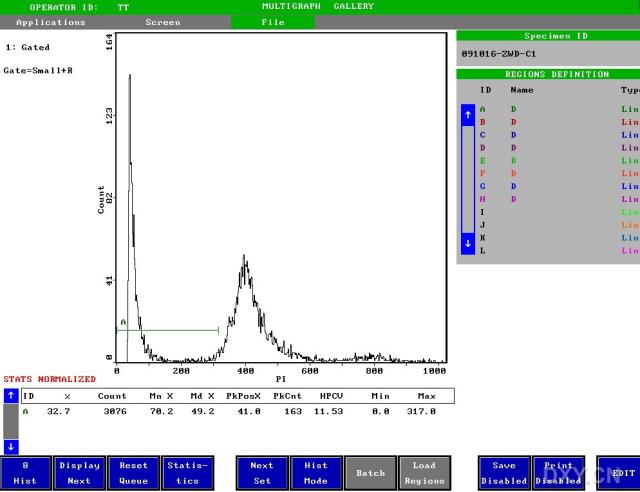 图片附件: 14492377.snap.jpg (2012-1-31 12:41, 39.49 KB) / 该附件被下载次数 8
图片附件: 14492377.snap.jpg (2012-1-31 12:41, 39.49 KB) / 该附件被下载次数 8 图片附件: 29055701.snap.jpg (2012-1-31 12:42, 53.97 KB) / 该附件被下载次数 8
图片附件: 29055701.snap.jpg (2012-1-31 12:42, 53.97 KB) / 该附件被下载次数 8 图片附件: 54925774.png (2012-1-31 12:43, 21.68 KB) / 该附件被下载次数 11
图片附件: 54925774.png (2012-1-31 12:43, 21.68 KB) / 该附件被下载次数 11
| 欢迎光临 分析测试百科 (http://bbs.antpedia.com/) | Powered by Discuz! 5.5.0 |