
查看完整版本请点击这里:
经验:钝峰分析,得出的解决方面与结果!!
1.产生钝峰的原因有哪些?经验:钝峰分析,得出的解决方面与结果!!
2.怎样避免钝峰,有何措施?
3.图中除了钝峰,还有随着保留时间的延长,基线上飘。有什么办法可以减少漂移?
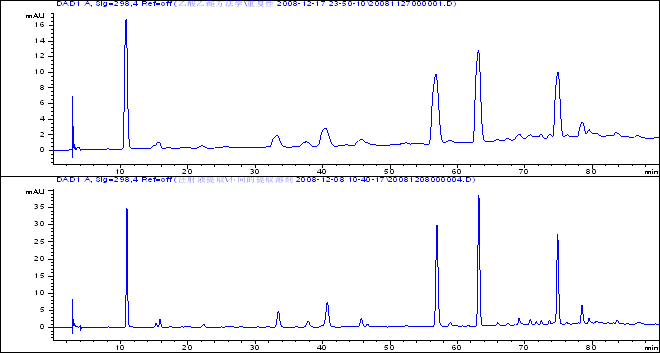
上图为钝峰图谱,下图为正常状态下的图谱,条件一样【梯度洗脱,甲醇:酸水(乙酸),298NM,ODS柱】
1.造成漂移的原因是电源电压不稳;温度及流动相流速的缓慢变化;固定相从柱中冲刷下来;更换的新溶剂在柱中尚未达到平衡等
通过加电源稳压器和接地措施、温度精确控制、恒压恒流装置来改善
2.如果条件没变化,最大的可能就是柱效不行了。
可以从图谱看出的是,峰高相差比较大,峰宽明显增加,保留时间没有明显变化。那么就是柱子的问题了。至于基线上漂的问题,我猜测是运行时间太长,柱子需要重新清洗,或者还是色谱柱使用时间太长了。
3.柱效下降往往导致保留时间的改变和峰形的改变
图中虽然峰宽增大,但是保留时间和峰形仍旧很好
推测可能是紫外检测器的扫描速率设定的过低,远大于半峰宽导致钝峰
基线漂移可以适当增加狭缝宽度
4.我觉得从两方面考,一是气体流量的稳定性,二是柱子污染或者是柱效下降。
5.肯定跟柱子是没有多大关系的,因为保留时间几乎是完全一致的。
现在需考虑的问题可能是检测器的问题,比如紫外灯的能量,波长的准确度与精密度需坐下验证。
6.温度是否稳定,有可能是灯的能量问题。
7.一定是紫外扫描频率被认为改过, 上图的扫描频率明显要低,导致在同样时间内,采集到的数据不足,形成钝缝,而且两张图的标尺明显不同,钝峰的浓度明显偏小,这也是造成钝峰的一方面原因.
8. 从图谱上看,两幅图中的保留时间基本一致,但是峰高差别几乎1倍,第一图比第二图出峰宽。结合斑竹提供分析条件,认为
问题1. 柱效降低;检测器的灵敏度小;流动相的pH值有较小的差异;
问题2. 更换新柱或老柱降低进样量;提高检测器的灵敏度或采样频率;使每次更换流动相的pH值保持一致或组成差别小。
问题3. 通常在梯度洗脱中不易根除基线的漂移,但是可以通过稳定环境温度来减小漂移;通过检测器或色谱工作站的空白背景扣除功能,来减去或扣除空白流动相的背景值或色谱图,来减小基线漂移的影响。
9. 钝峰可能是色谱柱效不行,还有就是样品最后定容溶剂(上机)也有关系,漂移可能是因为走梯度没有完全平衡好.
10.解决上面图的问题,首先彻底清洗柱子,因为基线噪音大,出现顿峰这是常见原因;其次要保证流动相比例的稳定和柱温的稳定
11.1.紫外灯能量可能降低了。2.柱效下降。
12. 偷偷看了一下两张谱图的时间,发觉出现钝峰的谱图时间是在10小时以后的,而且流动相是甲醇:乙酸,长时间进样会使柱子“劳损”,柱效降低,所以会出现钝峰。
个人认为消除这种现象,可考虑在进样一段时间后,用较高浓度甲醇冲柱,之后再继续进样,也就是在序列里加进一针冲柱的。(不过要注意:冲柱的方法文件一定要保存到序列文件夹里,否则会出错停机!1200跟1100一样,很笨的)
13.出钝峰:指所出的样品峰不尖,所有峰或一部分峰的顶部呈不规则形状(平头或园形)。
出现的原因:
A.进样量太大使色谱柱或检测器形成饱和,减少进样量或降低样品浓度。
B.进样器是否存在漏液现象。
C.检查分析条件的设置是否正确。
D.尝试提高进样器、检测器的温度,改善峰的形状。
14.在气相的毛细管柱上,即使是柱子用了很长时间的(时间一年多,做样3000组,进样10000次以上的柱子,柱流失已经非常大),只要是不截柱子,保留时间变化是很小的。
但是在液相则不同,进样超过5000次以上,或者在样品比较脏的情况下,过一段时间后,保留时间就会有所变化。
因为我的结论也是样的钝锋的问题如果是液相出现的话,应该不是柱子的问题。
15.分析图谱发现峰的保留时间不变,但峰整体展宽.我认为原因应该是体系的死体积变大了.可能的影响区域是
1.柱子接头处,各管线接头处未完全接合,造成死体积过大.
2. 进样的六通损坏.
解决办法是:
1.采用合适的接头和正确的接法使系统死体积最小.解决基线漂移的办法是将液相条件稳定.如柱温,压力等.
16.我觉得是与纵坐标的电压显示量程范围有关,上图为显示到16,下图为35,上图相当于还有峰的上半部分被挡在了显示屏外。总的来说,是放大的比例不同,所以显示出的细微差别不同 ,上图就是纵向放大了之后,看到的峰下面部分。
18.条件没有改变,主要是色谱柱的柱效下降引起。
19.应该是柱效降低造成的。主要原因是流动相比例或PH不合适,或者水相比例过大且长时间冲洗造成的,用有机相较高的溶剂或纯有机相慢慢冲洗柱效可以恢复。
20.从图看:柱效下降很多,看来先要彻底的冲洗柱子.流动相里有乙酸,做样前多平衡柱子,另外加上柱温箱漂移会好些.
21.可能是柱效降低引起钝峰,可换根较好柱子。
基线漂移不是很大,应没多大问题,改善基线漂移方法:流动相尽量用色谱纯,水用超纯水(如Millipore); 如果可能,可适当提高检测器波长(必须对分析结果影响不大),基线漂移不所改善。
22.基线向上漂移通常是由于所用A与B溶剂的紫外吸收差异所致。反相梯度洗脱中,有机溶剂B浓度在分离期间是增加的,有机溶剂的紫外吸收总是大于水,so,采用紫外检测器时,这种漂移尤为普遍。与吸收度相关的飘移可通过“吸收匹配”来消除(即在A溶剂中加入uv吸收化合物,以增加A溶剂的吸收,使与B溶剂相等)
23.可能是柱子或流动相的问题,更换一下试试
24.我觉得应该是检测器的问题。
溶剂条件不变,波形不应该变化
峰的保留时间基本不变
所以可能处在检测器上。
25. 如果仅是自外检测器的扫描速率设定过低,出峰时图谱不平滑,当不至于导致峰宽如图中这样增大。考虑到其保留时间和峰形仍旧完好,还是觉得柱效下降的可能性多一点.
[ 本帖最后由 miracle 于 2010-5-25 11:15 编辑 ]
查看完整版本请点击这里:
经验:钝峰分析,得出的解决方面与结果!!
经验:钝峰分析,得出的解决方面与结果!!

最新回复
sacred (2010-5-25 10:24:30)
27. 这种现象是否经过重复?还是偶然现象?这个首先要确定一下!另外好好清洗色谱柱和系统,看是否能消除?还有测样时系统流速、压力、温度是否正常?更换新的溶剂是否会改善?色谱的原因是不太好找的,要一点点排查。
28.我碰到过等能量减弱但是作出的峰好像还是挺对称的,只不过是峰面积小,所以我认为这不是主要原因吧。两个峰型差那么多,样品浓度和柱效以及温度恒定与否是主要考虑的原因。
29.个人认为检查一下管路是否脏了,保护柱是否需要更换,最好检查灯能量,是否需要更换了
30.我认为操作者不会去更改采集数据的频率的。
从图谱上可以看出,保留时间一致,因此pH和温度估计都没有改变。我推测有两种可能:1:柱子有点脏了,或使用时间比较长了。2:进样体积大了。
31.1、可能是柱子被污染了建议冲洗。2、你是否有在线脱气的设备,在线混合有可能产生气泡的。
32.钝峰是柱效降低的缘故;
基线漂移是梯度洗脱造成;
看来梯度的最后应用有机溶剂多冲冲柱子再进下一个样。改善基线漂移可移考虑在甲醇相也适当加酸
33.原因较多,排除仪器及色谱柱方面的原因,可能有以下:
原因 解决方法
1.进样体积过大 用流动相配样,总的样品体积小于第一峰的15%
2.在进样阀中造成峰扩展 进样前后排出气泡以降低扩散
3.数据系统采样速率太慢 设定速率应是每峰大于10点
4.检测器时间常数过大 设定时间常数为感兴趣第一峰半宽的10%
5.流动相粘度过高 增加柱温,采用低粘度流动相
6.检测池体积过大 用小体积池,卸下热交换器
7.保留时间过长 等度洗脱时增加溶剂含量也可用梯度洗脱
8.柱外体积过大 将连接管径和连接管长度降至最小
9.样品过载 进小浓度小体积样品
34.峰钝了应该是柱效下降了,基线上飘还在正常范围内,仪器接地可以减小影响。
35.柱子脏了,应该清洗再生。如果柱子前面安装有在线过滤或预柱的可能清洗或更换了。看图似乎有些飘移,应该跟运行时间长或是平衡不好有关。
其它的应该没有什么问题……
36.产生钝峰的原因主要有:1.色谱柱柱效下降;2.色谱条件与被分析物不匹配;3.检测器参数设置错误.
采取的措施:1.冲洗干净色谱柱,并经常活化色谱柱;2.筛选较佳的色谱条件(主要是色谱柱和流动相的选择);3.检查检测器的参数设置.
基线上漂:如果是等度洗脱基线上漂,主要原因可能还是系统还没到平衡状态或者无法达到较好的平衡状态,其次就是流动相对色谱柱损害较大.如果是梯度洗脱,基线规则的上漂或下漂应该是系统正常的漂移,基本上很难避免.
37.保留时间完全一致,峰高大约降低一半,但峰宽增大。
这个问题与柱温、 流动相、梯度条件等变化无关,进样量也不可能过载,体积过载也几乎不可能,因为搂主在做“重复性”试验。柱效和检测器的采样频率会影响峰形,从做样的时间来看正常峰是12-8做的,出现顿峰是12-17,故极有可能是柱子用了一阵后柱效降低造成的。
38.可能是柱效降低和改善流动相配比。
39. 进样方式有误、柱效降低、检测池受到污染、管道被堵以及电压不稳等都有可能出现钝峰。色谱操作上一定要注意每一个环节,否则会出现意想不到的图谱。
40. 柱子污染或者是柱效下降
41.产生钝峰的原因有很多,包括我们自己都意识不到的方面,如:
仪器本身,色谱柱本身,流动相的配比等,我个人认为可能性大的地方有:
1.流动相配比有问题,不是有机系和水系的比,而是其他要求加入的少量的试剂;
2.色谱柱时间比较久了,柱效下降了,这就要更换色谱柱了;
3.色谱柱前段或预柱被污染了,这就要反向冲洗色谱柱,使之再生,预柱就拿去超声清洗即可。
基线漂移的原因:
1. 主要是基线还没有稳定就进样了,需要一定的时间稳定;
2.仪器本身的稳定性不好,如岛津的,走样时间久了也会漂移;waters、安捷伦的还好吧
3. 房间温度的上升或下降也会对其有轻微的影响
4.对于梯度洗脱来说,流动相的配比也很重要,要确定在每一时间段运行的时间和比例是否能让仪器准确的办到(仪器本身问题)。
42.就图谱来看,首先是柱效应该是降低了,才出现的钝锋,就处理一个柱子。其次就是基线漂移,有可能是平衡时间不够,现一个就是检测器的问题了。总的来说检测器出现问题不太多。
43.产生钝峰的原因:
1.进样量过大,柱过载了;
2.色谱柱柱效下降了;
3.流动相的有机相比例太少。
基线漂移的原因:
1.流动相还没平衡好;
2.仪器本身的稳定性不好;
3.温度的影响。
44.保护柱可能脏了,也就是说整体柱效下降了大多会出现这种情况
45.说柱效下降,但如何说明保留时间的重复呢?从上下两图对比,可以发现不光是钝峰的出现而且分辨率也明显下降了,基线也更加平滑了,本人还是觉得检测器采样频率不够或者灯能量下降或者采用了更宽的平滑窗口平滑谱图所致。
46.1.从图中看,钝锋产生的原因应该主要是柱效下降了,而且分离度也降低了.
2.冲洗或换柱子或保护柱了
3.基线漂移主要应该是梯度洗脱所致,可以消除溶剂空白或是在乙酸水液中加入吸收消除剂,另外乙酸换成磷酸应该基线效果会好点,截止波长小一些.
dragon5 (2010-5-25 10:55:44)
utek (2010-5-25 10:56:39)
eesa_dc_seein (2010-5-25 11:01:17)
zhaohaimi (2010-5-25 11:06:22)
michael_b_rex (2010-5-25 11:10:21)
经验:钝峰分析,得出的解决方面与结果!!