
查看完整版本请点击这里:
【求助】怎样提高目标蛋白吸附到DEAE-Sephrose Fast Flow介质的量
相关疾病:【求助】怎样提高目标蛋白吸附到DEAE-Sephrose Fast Flow介质的量
46XX性发育睾丸疾病
各位做蛋白纯化的高手好,我现在在纯化人血浆中的一种蛋白,分子量52kDa,等电点4.55,前处理采用还原剂打断杂蛋白的二硫键(不影响目标蛋白),气相二氧化硅吸附脂蛋白。
上柱子前的蛋白含量为7mg/ml,平衡液为50mM pH 7.5的Tris,在平衡液中增加NaCl盐浓度0.05M、0.1M、0.15M、0.2M梯度洗脱,通过活性测定和SDS-PAGE电泳发现0.05NaCl洗脱杂蛋白,0.1MNaCl洗脱目标蛋白。
存在的问题是未结合在介质上峰很大,里面也有不少目标蛋白,活性比0.1MNaCl洗脱峰的活性还高。
我已经尝试提高平衡液的pH到8.0,其余都不变,效果还不如原来,未结合峰的活性更大。该怎样改变条件才能提高目标蛋白在介质上的结合量呢?求各位前辈指点迷津,感激不尽。
以下是我的AKTA层析图谱,第一个是pH 7.5,第二个是pH8.0
查看完整版本请点击这里:
【求助】怎样提高目标蛋白吸附到DEAE-Sephrose Fast Flow介质的量
【求助】怎样提高目标蛋白吸附到DEAE-Sephrose Fast Flow介质的量
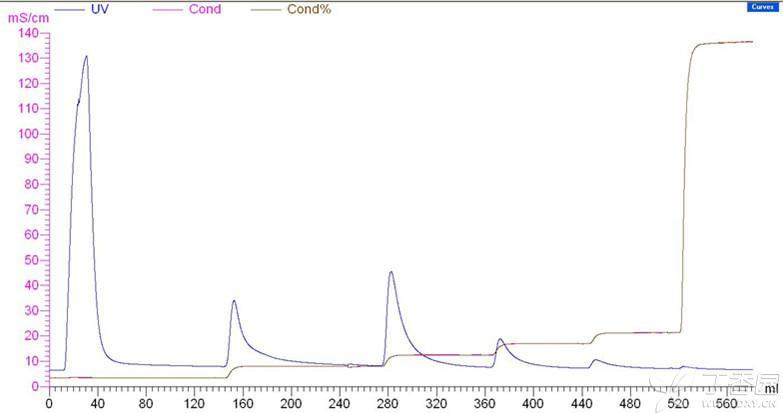
71301571.jpg

最新回复
兔唇 (2015-9-08 10:59:21)
这五个峰分别是未结合(峰很高)、0.05M、0.1M、0.15M、0.2MNaCl洗脱,后面是机器不稳定造成的峰
34602156.jpg
ROSE李 (2015-9-08 10:59:40)
我个人介意从以下几个方面入手:1、降低平衡液电导与样品电导。之前你用还原剂,样品中电导值必然会比较高。通过稀释降低电导值。2、提高上样pH,你也尝试了,可以进一步探索,在保证蛋白性质不改变情况下。3、降低流速,高流速下,结合性能下降。3、更换介质,DEAE属于弱阴离子交换,可以换成强阴离子试试。你的基架,琼脂糖的,可以换成其他种类,推荐你可以试试 ABI的POROS Q
兔唇 (2015-9-08 11:00:08)
QUOTE:
根据你提的几点建议我是这样想的:1.我没测过前处理上样前样品的电导率,也没透析,因为是预实验,就想大致看一下,最好是样品能处于平衡液那种状态下,吸附最准了,下次在做时决定透析一下,置换溶解蛋白的溶液为平衡液。2.提高平衡液的pH,我以前用试管法(介质装在试管中,不断用平衡液洗涤平衡,吸取上清液,再加平衡液洗涤,最后加杨平,测上清液的活性和蛋白含量)试过目标蛋白在pH7.0,7.5,8.0,8.5条件下,哪个吸附效果最高,结果是随着pH值的升高,上清液的活性也升高,说明目标蛋白没吸附上,pH7.0,7.5吸附情况差不多,这次用AKTA试果然pH 8.0效果不如7.5,所以要不要降低pH,换Tris为PBS?3.我的流速是2ml/min,上样和洗脱都是这个速度,还要降低吗?4.文献中都用的DEAE这个胶,或者是比较几种胶的效果,发现还是DEAE弱阴离子胶好一点。啰嗦了这么多就是不知道下一步该怎么办,实验卡住了,希望找个师兄师姐好好讨论一下,再次感谢你
ROSE李 (2015-9-08 11:00:41)
1、透析或者用超滤离心管换液至平衡缓冲液,可以有效降低样品电导值。2、既然pH你已经做过实验了,影响不大,那平衡液的电导值呢? 50mM的浓度你是怎么得来了,一般都会产用20mM作为起始离子强度,若结合效果好,可再进一步优化。既然你为了保证不穿透,可以尝试降低你的平衡液浓度,10mM 20mM均可。3、 流速,你给我的是体积流速,你柱子是多少尺寸的,你可以自己换算下,推荐线性流速100cm/h时,看有木有增加你的结合量。4、缓冲液阴离子一般就用Tris 其PK大概在8.0左右,缓冲效果好,无需更换成PB
兔唇 (2015-9-08 11:01:16)
QUOTE:
1.周三做过一次用超滤离心管换平衡液的实验,超滤后35ml体积变成30ml,蛋白含量由4mg/ml降低到2mg/ml,不过比活没有下降太多,证明可行。不过这次的平衡液我用了0.05M pH 6.5的PS,AKTA层析图的未结合峰降低了很多,还以为改变条件结合上去了,但测活的结果显示未结合峰依然活性很高。2.起始缓冲液的浓度是看纯化该蛋白的专利得到的,它有个范围,我就选了一个中间的浓度,感觉20或者10mM是不是太低了,这样缓冲液的缓冲能力比较小,体系不够稳定,50mMTris的离子强度是2.2左右,周三PS的离子强度为4.2。3.我用的是GE的小柱子,直径1.6cm,这几次次我都装15ml胶左右,它的载量大约110mg/ml,算了一下,线性流速为60cm/h。4.换了缓冲溶液效果并不好,今天的实验改用Tris在做一次。出峰分别为未结合、0.05MNaCl、0.1MNaCl、0.5MNaCl 纵坐标是电导率,截图时忘了换成UV
0.5MNaCl洗出两个峰,可能是盐浓度一下子增加太多引起的
这是样品的实验数据
57606662.png
ROSE李 (2015-9-08 11:01:39)
图谱的值下降,源于你样品浓度低了。50mMTris的离子强度是2.2左右,周三PS(你用的是PB还是PBS ?)的离子强度为4.2,相通浓度的,PB的离子数更多,虽然只差2.2,但对离子交换的影响是巨大的额,我之前仅差0.5单位,样品回收率就差很多。50mMTris的离子强度不大,与20mMpb相当,不需要换。
关于缓冲能力,浓度越低,缓冲能力下降,但是由于你样品也在平衡液体系中,期间不会有过强酸碱引入,不会造成pH波动的,我们这所有的平衡液体系都是20mM,我看技术手册也都是20mM的体系,升高平衡液pH有可能使结合不牢的杂蛋白去除,当然也会使目的蛋白结合力下降。 介质载量110mg/ml,是理论的,特定的条件的,实际远远低于,但是你用15ml的 柱子,,应该也不会超过它载量的,。。。很奇怪。。 请问用完之后有CIP么?
ssonglikihi (2015-9-08 11:01:57)
是只想得到较纯的目的蛋白做分析呢?纯度有何要求?
还是需要优化工艺提高收率和分辨率?
如果是后者,选择一个合适的填料是首要的,主要考察其结合能力、分辨率,可尝试一下强阴的Q;其次是筛选一个合适的上样条件,离子交换也就是筛选合适的上样pH,阴离子结合洗脱模式一般高于等电点0.5个pH以上,当然要注意上样时电导率低一些(一般<5 mS/cm);在填料和上样条件确定的情况下,就要去测你目的蛋白的动态结合载量(DBC),这样,你才能确定你的上样量,避免流穿;洗脱可用线性梯度洗脱来提高分辨率和收率,再由线性洗脱条件转化为阶梯洗脱,以节约时间和Buffer。
根据你的情况,建议降低上样量,看是否还会有流穿,用盐浓度线性梯度洗脱,0-20%B (1 M NaCl),20 CV,分管收集看主要成分在哪里。
兔唇 (2015-9-08 11:02:27)
QUOTE:
介质用完用1MNaOH洗,再换2MNaCl,再换20%乙醇,用的是说明书里的再生方法,没用用在位清洗。周五做的实验还是原来的条件pH 7.5Tris平衡,0.05MNaCl洗脱杂蛋白,0.1MNaCl洗目标蛋白,只在上样之前增加了超滤管超滤,目标蛋白就结合上去了,AKTA图谱上0.1MNaCl峰达到1400mAU,未结合不到400mAU,测活是光看颜色就知道目标蛋白结合上去并被洗脱下来了,这次的结果出乎意料,很理想,原来只是要增加一步超滤就可以解决问题啊,我又重复了一次,没周五的结果好,但基本上重复出来了,非常感谢cpu随风而逝提的建议,作出结果我就回家休息了一段时间,刚回来进行第二步精制纯化,再次感谢。
兔唇 (2015-9-08 11:03:11)
QUOTE:
我的课题就是把目标蛋白纯化出来,纯度要达到95%,对蛋白进行鉴定。填料是组长定的,刚到实验室对胶也不熟悉,现在恶补了一点专业知识才好一点。
在上样前增加一步超滤,目标蛋白已经可以结合上去了,平衡液为pH 7.5的tris,离子强度2.2,满足低电导率的要求。
我是初步摸索15ml胶,20ml样品,浓度为3mg/ml,上样量是比较小的,这样做的快,利于分析。
不过最终还是要放大,你提到的动态结合量相信对我有很大的帮助,现在还不知道怎么测,不过我会好好学习一下,确定上样量,不懂的地方再来请教你
【求助】怎样提高目标蛋白吸附到DEAE-Sephrose Fast Flow介质的量